Famotidine Tablets
Action and use
Histamine H2 receptor antagonist; treatment of peptic ulceration.
Definition
Famotidine Tablets contain Famotidine.
Content of famotidine, C8H15N7O2S3
95.0 to 105.0% of the stated amount.
Identification
TESTS
Dissolution
Comply with the requirements for Monographs of the British Pharmacopoeia in the dissolution test for tablets and capsules, Appendix XII B1, using Apparatus 2. Rotate the paddle at 50 revolutions per minute and use as the medium 900 mL of phosphate buffer pH 4.5 prepared in the following manner. Dissolve 13.61 g of potassium dihydrogen orthophosphate in water, adjust the pH of the solution with orthophosphoric acid or 1m potassium hydroxide as necessary and add sufficient water to produce 1000 mL. Withdraw a sample of 10 mL of the medium, filter and centrifuge at 2000 revolutions per minute for 20 minutes. Carry out the method for liquid chromatography, Appendix III D, using the following solutions. Solution (1) contains 0.001% w/v of famotidine BPCRS in the dissolution medium. For solution (2) use the filtered and centrifuged dissolution medium, diluted if necessary, to give a solution expected to contain about 0.001% w/v of Famotidine.
The chromatographic procedure described under Related substances may be used. Inject 50 µL of each solution.
Calculate the total content of famotidine, C8H15N7O2S3, in the medium using the declared content of C8H15N7O2S3 in famotidine BPCRS.
Related substances
Carry out the method for liquid chromatography, Appendix III D, using the following solutions. For solution (1) add 200 mL of 0.05m potassium dihydrogen orthophosphate adjusted to pH 6.0 with 1m potassium hydroxide (solution A) to a quantity of whole tablets containing 0.2 g of Famotidine, mix with the aid of ultrasound for 5 minutes, add about 200 mL of methanol, shake for 60 minutes and add sufficient solution A to produce 500 mL. For solution (2) dilute 1 volume of solution (1) to 100 volumes with solution A. For solution (3) dilute 1 volume of solution (1) to 10 volumes with solution A and dilute 1 volume of the resulting solution to 50 volumes with solution A. For solution (4) add 40 mL of acetonitrile to a mixture of 2 mg of each of famotidine impurity C BPCRS, famotidine degradation impurity 1 BPCRS and famotidine degradation impurity 2 BPCRS, mix with the aid of ultrasound for 2 minutes, cool, add 40 mL of methanol and sufficient solution A to produce 200 mL. Dilute 1 volume of the resulting solution to 5 volumes with solution A. For solution (5) dissolve 8 mg of famotidine BPCRS in 15 mL of solution A with the aid of ultrasound, cool and add sufficient solution A to produce 20 mL (solution B); to 1 mL of this solution add 0.05 mL of hydrogen peroxide solution (10 vol) (generates famotidine degradation impurity 3). For solution (6) dilute 1 volume of solution B with 100 volumes of solution A and further dilute a suitable volume with an equal volume of solution (4).
The chromatographic procedure may be carried out using (a) a stainless steel column (15 cm × 4.6 mm) packed with end-capped octadecylsilyl silica gel for chromatography (5 µm) (Inertsil ODS-2 is suitable) maintaining the column temperature at 40°, (b) a mixture of 7 volumes of acetonitrile and a mixture of 93 volumes of 0.1m sodium acetate containing 0.1% v/v of triethylamine the pH adjusted to 6.0 with glacial acetic acid and as the mobile phase with a flow rate of 1.4 mL per minute and (c) a detection wavelength of 275 nm. Condition the column for approximately 30 minutes before the first injection.
Inject separately 50 µL of each solution. The substances are eluted in the following order: famotidine degradation impurity 3, famotidine degradation impurity 1, famotidine impurity C, famotidine and famotidine degradation impurity 2. The test is not valid unless, in the chromatogram obtained with solution (6), the resolution factor between the peaks due to famotidine and famotidine impurity C is at least 1.4 and that between the peaks due to famotidine and famotidine degradation impurity 2 is at least 1.4. If this resolution is not achieved adjust the content of acetonitrile or decrease the concentration of sodium acetate in the mobile phase.
In the chromatogram obtained with solution (1) the areas of any peaks corresponding to famotidine impurity C or famotidine degradation impurities 1 and 2 are not greater than the areas of the corresponding peaks in the chromatogram obtained with solution (4) (0.5% of each), the area of any peak with a relative retention time of about 0.4 with reference to famotidine in the chromatogram obtained with solution (5) (famotidine degradation impurity 3) is not greater than the area of the peak in the chromatogram obtained with solution (2) (1%) and the area of any other secondary peak is not greater than the area of the principal peak in the chromatogram obtained with solution (3) (0.2%). Calculate the contents of each of impurity C and degradation impurities 1 and 2 using the chromatogram obtained with solution (4) and the nominal content of any other impurities using the chromatograms obtained with solution (2) and (3). The sum of the contents of all the impurities so calculated is not greater than 2.5%. Disregard any peak with an area less than 0.25 times the area of the principal peak in the chromatogram obtained with solution (3) (0.05%).
Assay
Carry out the method for liquid chromatography, Appendix III D, using the following solutions. For solution (1) add 200 mL of 0.05m potassium dihydrogen orthophosphate adjusted to pH 6.0 with 1m potassium hydroxide (solution A) to a quantity of whole tablets containing 0.2 g of Famotidine, mix with the aid of ultrasound for 5 minutes, add about 200 mL of methanol, shake for 60 minutes and add sufficient solution A to produce 500 mL. Dilute 1 volume of this solution to 5 volumes with solution A. For solution (2) dissolve 8 mg of famotidine BPCRS in 4 mL of methanol, mix with the aid of ultrasound for 5 minutes and add sufficient solution A to produce 20 mL. Dilute 1 volume of this solution to 5 volumes with solution A.
The chromatographic conditions described under Related substances may be used. Inject 50 µL of each solution.
Calculate the content of C8H15N7O2S3 in the tablets using the declared content of C8H15N7O2S3 in famotidine BPCRS.
IMPURITIES
The impurities limited by the requirements of this monograph include impurity C listed under famotidine and the following degradation impurities.
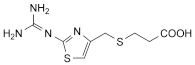
1. 3-[2-(Diaminomethyleneamino)-1,3-thiazol-4-ylmethylthio]propanoic acid,
Corresponds to impurity F listed under Famotidine
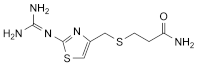
2. 3-[2-(Diaminomethyleneamino)-1,3-thiazol-4-ylmethylthio]propanamide,
Corresponds to impurity D listed under Famotidine
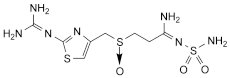
3. 3-[2-(Diaminomethyleneamino)-1,3-thiazol-4-ylmethylsulfinyl]-N-sulfamoylpropanamidine.