Appendix II C. Nuclear Magnetic Resonance Spectrometry
Introduction
Nuclear magnetic resonance (NMR) spectrometry is an analytical method in particular suitable for the elucidation of the chemical structure of organic molecules by means of interpretation of their NMR spectra, arising from, for example, 1H or the X-nuclei 13C, 19F, 15N, 31P. The spectra can be used for qualitative and quantitative purposes.
Under suitable experimental conditions, the integrated NMR intensities of the signals are directly proportional to the number of nuclear spins of the molecular group responsible for the signal. These integrals can be used for quantitative analysis.
GENERAL PRINCIPLES
Placing an ensemble of nuclei with angular momentum and a magnetic moment in a static magnetic field (B0) causes the nuclei to arrange themselves in different, quantum-mechanically controlled orientations in relation to the axis of the magnetic field. These orientations are different in energy. An oscillating high-frequency magnetic field (B1), perpendicular to B0, will cause transitions between these orientations with net energy absorption. According to the resonance condition ω0 = γB0 (γ = gyromagnetic ratio, ω0 = Larmor frequency), either the B0 magnetic field or the frequency (ω1) of the B1 field may be varied to achieve a spectrum (continuous wave (CW) method). Nowadays the B1 irradiation is achieved by the use of a radiofrequency (RF) pulse (Fourier transform (FT) method). The coherent radiation emitted during the return to the initial state is observed in the form of a decay curve, called the free induction decay (FID). Subsequent Fourier transformation gives the spectrum in the frequency domain, providing information about the molecular structure. Additional radiofrequency fields may be applied during acquisition of the FID signal to suppress scalar (through-bond) interactions between nuclei (called ‘decoupling’). One- and multi-dimensional techniques can be applied for qualitative and quantitative purposes, on samples in either the liquid or the solid state.
Important structural information is derived from the following spectroscopic features:
Correlations of different spectral parameters (e.g. chemical shift and coupling constant, or chemical shifts of different nuclei within one molecular system) can be performed by homo- and hetero-nuclear two- and higher-dimensional methods. Information about the relaxation times T1 and T2, nuclear Overhauser effects (NOEs) and the kinetics of time-dependent processes are also accessible from appropriate experiments.
APPARATUS
A high-resolution NMR spectrometer consists of at least the following parts:
and may also include:
The high magnetic field is generated by a superconducting coil in a Dewar flask filled with liquid helium. The probe typically contains the sample in a 5 mm-outer-diameter sample tube or in a flow cell, and is connected to the electronics cabinet by RF cables carrying lock, 1H-, and X-nucleus frequencies. Additional devices for tuning and matching the electronic circuits are essential, and sample temperature control is often used.
The NMR spectrometer should be demonstrated to be operating correctly. Appropriate tests to demonstrate this are, typically, measurement of linewidths at half height for defined peaks under defined acquisition conditions, signal-to-noise ratios (S/N) for standard mixtures, pulse power (measured as a 90° pulse width), and pulse reproducibility. All instrument manufacturers publish specifications and measurement protocols for these parameters for specific instrument/probe combinations, and compliance with these specifications should be demonstrated.
FOURIER TRANSFORM NMR (FT-NMR)
Contemporary spectrometers generally operate according to the Fourier transform (FT) principle: after exciting the sample with a radiofrequency pulse of appropriate frequency (ν), amplitude (B1) and duration (τp) and a succeeding short dead time (td) (to enable the electronics to recover), the amplified analogue FID signal is sampled during the acquisition time (tac) and digitised with an analogue-to-digital converter (ADC), and the results are stored in the spectrometer memory. The receiver output is amplified prior to digitisation to maximise sensitivity without saturating the ADC. In case of observation of X-nuclei, the standard experiment includes, if necessary, broadband 1H decoupling, i.e. irradiation of all the protons during the experiment. To increase the S/N, multiple FID signals may be accumulated coherently and summed. Fourier transformation of this time-domain data gives the frequency-domain spectrum.
PARAMETERS
The following acquisition parameters influence the result of an FT experiment, and should be adjusted and controlled.
Pulse width (τp)
The excitation pulse is directed along the x-axis of the so-called rotating frame, its duration (or ‘width′, τp) determines the flip angle (θ) and thus the intensity (I) of the resonance signal:
The observed magnetisation My is maximum at θ = 90°. The pulse duration should be short to guarantee that all signals in the spectral width (SW) are excited to a similar degree. The magnetisation decays due to relaxation processes.
Dead time (td)
The dead time is the time between the end of the pulse and start of the acquisition, it is necessary for technical reasons and care should be taken as it may influence signal intensities and peak phase. Rapidly decaying signals (giving rise to broad spectral lines) are reduced in intensity by more than slowly decaying signals (which give rise to narrow spectral lines).
Acquisition time (tac)
The acquisition time (tac) is related to the spectral width (i.e. the whole observed region) and the number of digital data points (DP) collected during signal acquisition.
Maximal signal intensity and signal-to-noise ratio will be achieved if tac ≈ 1.2/(πν1/2), where ν1/2 is the full width at half-height (fwhh), but it should be set to greater than 5/(πν1/2) to minimise signal distortion.
Repetition time (tr)
The spin-lattice relaxation (T1) governs the time required for the spin system to return to equilibrium after a pulse. Relaxation can be reduced by the use of special reagents. For quantitative purposes, the repetition time used should be set relative to T1 and θ to avoid saturation effects.
Receiver gain
The analogue signal detected by the probe is amplified prior to digitisation and storage. The amplification, or receiver gain, should be set, either automatically or manually, so that the signal does not overload the ADC, which causes signal distortion, but allows random noise generated in the probe to be digitised (i.e. is non-zero).
OPTIMISATION OF ACQUISITION and processing PARAMETERS FOR QUANTITATIVE PURPOSES
Besides the acquisition parameters, signal intensity is also influenced by several processing parameters. After collecting a sufficient number of scans, the resulting FID is Fourier transformed. For reliable quantitative purposes the following parameters have to be optimised.
Digital resolution
The digital resolution is the frequency separation between data points. The processed signal should have at least 5 digital points above half-height of the signals to be integrated. To improve the digital resolution additional points of zero intensity may be added to the end of the experimental FID before transformation (‘zero filling’).
Signal-to-noise ratio (S/N)
This is the ratio between the intensities (as peak height) of a specified signal in the NMR spectrum and the random fluctuations in that signal, which is usually measured in a region of the spectrum that contains no signals from the analyte. A poor signal-to-noise ratio (S/N) limits the accuracy of peak integrations and quantitative analyses. An S/N equal to or greater than 150:1 allows peak integrations with a standard deviation of less than 1 per cent. Contemporary spectrometers have software algorithms to report the S/N of appropriate peaks. A sufficiently high S/N can be difficult to obtain when analysing dilute solutions, and especially when detecting nuclei other than 1H. Methods to enhance the S/N include:
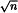 ;
;Integration region
The intensity of the NMR signals is obtained by a quasi-analogue signal integration either by a stepped-line plot or, more accurately, by separate line integration and digital data presentation. In liquid state, NMR signals have Lorentzian line shape. Unless otherwise specified in the monograph or when peak overlap occurs, the same integration range, expressed as a multiple of the peak fwhh, should be used for the monitored peak and the reference peak.
Dynamic range
The dynamic range of the analogue-to-digital converter (ADC) determines the minimum intensity line that can be observed or quantified when integrating 2 signals with the same linewidth in a spectrum. A 16-bit ADC allows identification of a signal of 0.003 per cent intensity relative to a strong signal completely filling the dynamic range of the ADC.
NMR OF SAMPLES IN SOLUTION
Most NMR experiments are performed on dilute solutions (about 1 per cent) of the analyte in an appropriate solvent, which can be spiked with a suitable standard for chemical shift calibration.
Solvents
The solvent should be able to dissolve the analyte without further interaction if not otherwise intended. To minimise the intense solvent signals, fully deuterated solvents (deuterium oxide R, deuterated chloroform R, deuterated dimethyl sulfoxide R, deuterated acetone R, deuterated methanol R, etc.) should be used. The solvent atoms give signals that are easily identified by their chemical shift and can be used to calibrate the chemical shift axis (secondary reference).
Referencing
The spectral feature most dependent on the chemical environment of the atom in the molecule is the chemical shift, designated as δ and reported in parts per million. The chemical shift between the resonance for an NMR active nucleus X (δX,sample) is measured in parts per million as the difference between the resonance frequency of that nucleus (νX,sample) and that of an internal shift reference standard (νX,reference), both in hertz, divided by the basic spectrometer operating frequency (νX,reference), in megahertz, at a given B0:
By convention, the standard for exact chemical shift referencing is the 1H resonance of tetramethylsilane R (TMS), setting δTMS = 0 ppm. Formally, once the 1H shift scale has been referenced relative to TMS, the exact frequency of any other X resonance can be calculated and its chemical shift scale calibrated. The frequency of a (secondary) reference standard at δX = 0 ppm (νX,reference) is calculated from the 1H frequency of TMS (νH,TMS) and a tabulated value of the ratio (ΞX,reference) of the isotope-specific frequency in relation to that of 1H in TMS:
Reference standards at δX = 0 ppm and corresponding ΞX,reference values are shown below:
In practice, X chemical shifts are referenced directly using an appropriate standard. In 1H and 13C NMR, internal referencing is mainly used, where the reference is added directly to the system under study. In 15N, 19F and 31P NMR, external referencing is often suitable, involving sample and reference contained separately in coaxial cylindrical sample tubes.
Lock
In order to prevent drifting of the spectrum over time, a stabilising procedure, called field-frequency locking, is performed. The 2H (deuterium) signal arising from deuterated solvents is used to achieve this, unless otherwise specified in the monograph.
QUALITATIVE ANALYSIS
The principal use for qualitative NMR spectra is as an identity test, in which the 1H or 13C spectrum of a test sample is compared to the spectrum of a reference sample or, less commonly, with a published reference spectrum. Spectra of reference and test samples should be acquired using the same procedure and operational conditions. The peaks in the 2 spectra, or characteristic regions of the spectra, should correspond in position, intensity and multiplicity. In appropriate cases, mathematical comparison, such as calculation of a correlation coefficient, may be appropriate. In the absence of a reference standard, an in-house reference may be used, whose identity has been demonstrated by alternative methods, or the demonstration that the NMR spectrum is fully consistent with the reported structure for that material.
QUANTITATIVE ANALYSIS
Signal intensity in the basic NMR experiment is the integrated area under the signal curve measured. Only when 2 signals have equal fwhh and the same multiplicity may signal height serve as a measure of intensity. Under conditions of essentially complete relaxation between scans, the signal intensity (IA) is a true measure of the number (NA) of nuclei responsible for the respective signal:
The constant KS includes fundamental constants, properties of the sample and receiver parameters, and can be omitted in cases where signal intensities are compared, giving the direct relation between the numbers of nuclei in the 2 compared structure groups A and B:
The numbers (Ni) of nuclei belonging to different structure groups of 1 molecule are small integers. The values measured are rounded to the closest integer numbers. However, the proper operation of acquisition and processing of the spectrometer is easily checked by comparing exact intensities within a spectrum of any suitable organic compound of known structure.
In addition to the fact that the intensities of signals arising from each component in a mixture are related to each other by small integer numbers, the relative molar amounts of these components can be measured by comparison of the normalised intensities of resonances from different components. The molar ratio of 2 components of a mixture is calculated according to the following equation:
The determination is only valid in cases where the structure of the molecules for which IA and IB are determined are known (or at least the values of N for the monitored groups). Determinations are made using either an internal standard method or a peak-normalisation procedure.
Internal standard method
The mass (mA) of an analyte (A) can be determined if a known mass (mB) of a substance (B) with a known percentage content (PB) is added to the solution as an intensity standard. Equation (8) can be converted to equation (9):
Here, Mi are the molecular masses.
The intensity standard has to be carefully chosen; it should be completely soluble in the solvent used for the analyte, should produce only a small number of signals, and the ‘monitor group’ should have a signal in an empty spectral region. A compound of high purity and with a relatively high molecular mass is recommended for this purpose.
Normalisation procedure
The relative proportions of components in a mixture, the degree of substitution in a structurally modified polymer, or the amount of a contaminant can be determined by comparison of the relative intensities of resonances present.
The experimental method should be validated to ensure that there is no overlap of the relevant signals. When the contaminant is of poorly defined structure or molecular mass (e.g. an emulsifier), addition of known amounts of that material to the NMR tube will allow a calibration curve to be constructed.
METHOD
Sample handling
Dissolve the sample in the solvent to which the appropriate reference material may have been added to calibrate chemical shift, as prescribed in the monograph. For quantitative analysis, the solutions must be free from solid particles. Some quantitative analyses may require an internal standard to be included, so that integrations of resonances from the test sample and the reference material can be compared. Appropriate references and concentrations are indicated in the specific monographs. In other quantitative analyses, the result is obtained by comparing the relative intensities of 2 or all of the resonances that arise from the test sample. After loading the sample into a tube and capping, the sample is introduced into the NMR magnet, the experimental parameters are loaded and the experiment is executed. Key experimental parameters are indicated in the monograph.
The measurement procedure
Equilibrate the sample in the probe, and optimise the instrument to achieve best resonance conditions and to maximise the S/N by tuning and matching the probe, and make adjustments to maximise magnetic field homogeneity over the sample volume (called ‘shimming’). Record, or save to computer, the parameter settings. An experiment may be composed of multiple pulse-acquisition-delay sequences, and the individual FIDs are summed in the computer memory, with random noise being averaged out. When an appropriate S/N has been achieved, the FID is stored and the frequency-domain spectrum is generated by Fourier transformation of the summed FIDs.
NMR IN THE SOLID STATE
Samples in the solid state can be analysed using NMR spectrometers specially equipped for that purpose. Certain technical procedures make observable individual lines for individual atomic sites with a valuable extension of the applicability of NMR to inorganic materials as well.
One technique is the rapid rotation (4-30 kHz) of the powdered sample in a rotor (about 4 mm outer diameter) inclined at an angle of 54.7° (the ‘magic angle’) to the direction of the B0 magnetic field axis. This technique is named magic angle spinning (MAS). Another effective tool is high-power decoupling and a 3rd method is the transfer of polarisation from easily excitable nuclei towards less-polarisable nuclei, i.e. cross polarisation (CP). The combination of these techniques makes available high-resolution spectra containing much information about chemical and structural details in solid glassy, amorphous, and crystalline materials of ceramic, polymeric or mineralogical origin.
If NMR is applied to a solid, full details of the procedure are provided in the monograph.