|
|
|
|
|
|
|
1. Analogous and Nonanalogous Data Parameters |
|
|
|
|
|
|
|
|
Numerous variables have been derived from in vivo and in vitro data that have been correspondingly correlated with in vitro and in vivo data, respectively, as illustrated in Table 7. Several investigators have argued over the relative importance of different in vitro dissolution parameters. However, it appears that the time for 50% of the drug to dissolve in vitro (T50) is probably the best in vitro variable to correlate. Two factors that contribute to this observation are |
|
|
|
|
|
|
|
|
1. By employing this value, one is not limiting or biasing the observation to any formal kinetic interpretation of data. |
|
|
|
|
|
|
|
|
2. The T50 value indicates the central tendency of the in vitro dissolution performance. |
|
|
|
|
|
|
|
|
It has been pointed out that instead of confining oneself to a specific observation point (value), it is more optimal to consider the entire dissolution profile from T = 0 to T = 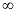 or some other parameter derived therefrom. This parameter often provides for a better and more meaningful indication of how the dosage form will perform in vivo. or some other parameter derived therefrom. This parameter often provides for a better and more meaningful indication of how the dosage form will perform in vivo. |
|
|
|
|
|
|
|
|
The use of areas derived from plasma concentration time profiles involves the assumption that plasma clearance of drug is the same following different treatments. Additionally, the amount of drug absorbed per milliliter of the volume of distribution varies with the pharmacokinetic model applied for analysis. Use of this parameter involves assumptions such as assuming that the volume of distribution is the same following different treatments. Additionally, the estimation of the elimination rate constants after different treatments is not biased by measurement in a region where absorption is either incomplete or in progress. However, in such instances when elimination rate constant is estimated, absorption has ceased. Such differences and/or inconsistencies should be taken into consideration and thoroughly understood before any inferences are drawn. |
|
|
|
|
|
|
|
|
The variables cited in Table 7 are meaningful in terms of the pharmacokinetic theory. It must be pointed out that estimation of relative bioavailability determined from parameters such as the amount excreted at a specific time, t, especially when t is less than ten biological-half-lives of the drug, cannot be employed for correlation. Also, area-under-the-plasma-concentration-time curve values of truncated blood-level-profiles when following concentration are measurable following one or multiple dosing and are inappropriate and should not be employed in correlating in vitro-in vivo data. The resultant correlations may not be valid in addition to the fact that they have less meaningful value. |
|
|
|
|
|
|
|
|
Quantitative correlations of appropriate variables derived from in vitro dissolution data with appropriate variables derived from in vivo performance of the dosage form in human subjects must be established and demonstrated in |
|
|
|
|
|