Appendix X N. Fixed Oils
Alkaline Impurities in Fatty Oils
In a test-tube mix 10 mL of recently distilled acetone R and 0.3 mL of water R and add 0.05 mL of a 0.4 g/L solution of bromophenol blue R in alcohol R. Neutralise the solution if necessary with 0.01 M hydrochloric acid or 0.01 M sodium hydroxide. Add 10 mL of the oil to be examined, shake and allow to stand. Not more than 0.1 mL of 0.01 M hydrochloric acid is required to change the colour of the upper layer to yellow.
Identification of Fixed Oils by Thin-layer Chromatography
Method A
Thin-layer chromatography (2.2.27).
Test solution Unless otherwise prescribed, dissolve about 20 mg (1 drop) of the fatty oil in 3 mL of methylene chloride R.
Reference solution. Dissolve about 20 mg (1 drop) of maize oil R in 3 mL of methylene chloride R.
Plate A suitable octadecylsilyl silica gel for high performance thin-layer chromatography as the coating substance.
Application 1 µL.
Development Twice over a path of 0.5 cm with mobile phase A, then twice over a path of 8 cm with mobile phase B.
Drying In air.
Detection Spray with a 100 g/L solution of phosphomolybdic acid R in ethanol (96 per cent) R. Heat the plate at 120 °C for about 3 min and examine in daylight.
The chromatogram obtained typically shows spots comparable to those in Figure 2.3.2.-1.
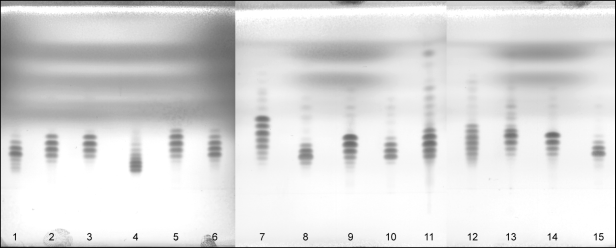
1. arachis oil | 4. rapeseed oil | 7. linseed oil | 10. almond oil | 13. evening primrose oil | |
2. sesame oil | 5. soya-bean oil | 8. olive oil | 11. wheat-germ oil | 14. safflower oil (type I) | |
3. maize oil | 6. rapeseed oil (erucic acid-free) | 9. sunflower oil | 12. borage oil | 15. safflower oil (type II) |
Method B
Thin-layer chromatography (2.2.27).
Test solution Unless otherwise prescribed, dissolve about 20 mg (1 drop) of the fatty oil in 3 mL of methylene chloride R.
Reference solution Dissolve about 20 mg (1 drop) of maize oil R in 3 mL of methylene chloride R.
Plate A suitable octadecylsilyl silica gel for high performance thin-layer chromatography as the coating substance.
Mobile phase methylene chloride R, glacial acetic acid R, acetone R (20:40:50 V/V/V).
Application 1 µL as bands of 8 mm. A suitable automated apparatus may be used.
Development Over a path of 7 cm.
Drying In air.
Detection Treat with a 100 g/L solution of phosphomolybdic acid R in ethanol (96 per cent) R. Heat the plate at 120 °C for 3 min and examine in daylight.
The chromatogram obtained typically shows zones comparable to those in Figure 2.3.2.-2.
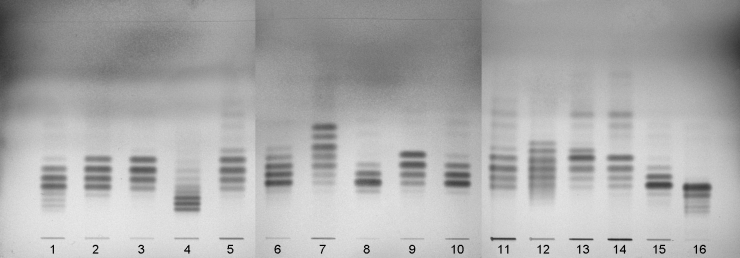
1. arachis oil | 5. soya-bean oil | 9. sunflower oil | 13. evening primrose oil | |
2. sesame oil | 6. rapeseed oil (erucic acid-free) | 10. almond oil | 14. safflower oil (type I) | |
3. maize oil | 7. linseed oil | 11. wheat-germ oil | 15. safflower oil (type II) | |
4. rapeseed oil | 8. olive oil | 12. borage oil | 16. hydrogenated arachis oil |
Test for Foreign Oils by Thin-layer Chromatography
Examine by thin-layer chromatography (2.2.27) using kieselguhr G R as the coating substance. Impregnate a plate by placing it in a chromatographic tank containing the necessary quantity of a mixture of 10 volumes of liquid paraffin R and 90 volumes of light petroleum R so that the plate dips about 5 mm beneath the surface of the liquid. When the impregnation mixture has risen by at least 12 cm from the lower edge of the plate, remove the plate and allow the solvent to evaporate for 5 min. Carry out the chromatography in the same direction as the impregnation.
Preparation of the mixture of fatty acids Heat 2 g of the oil with 30 mL of 0.5 M alcoholic potassium hydroxide under a reflux condenser for 45 min. Add 50 mL of water R, allow to cool, transfer to a separating funnel and extract with three quantities, each of 50 mL, of ether R. Discard the ether extracts, acidify the aqueous layer with hydrochloric acid R and extract with three quantities, each of 50 mL, of ether R. Combine the ether extracts and wash with three quantities, each of 10 mL, of water R; discard the washings, dry the ether over anhydrous sodium sulfate R and filter. Evaporate the ether on a water-bath. Use the residue to prepare the test solution. The fatty acids may also be obtained from the soap solution prepared during the determination of the unsaponifiable matter.
Test solution Dissolve 40 mg of the mixture of fatty acids obtained from the substance to be examined in 4 mL of chloroform R.
Reference solution Dissolve 40 mg of the mixture of fatty acids obtained from a mixture of 19 volumes of maize oil R and 1 volume of rapeseed oil R in 4 mL of chloroform R.
Apply to the plate 3 µL of each solution. Develop over a path of 8 cm using a mixture of 10 volumes of water R and 90 volumes of glacial acetic acid R. Dry the plate at 110 °C for 10 min. Allow to cool and, unless otherwise prescribed, place the plate in a chromatographic chamber, with a tightly fitting lid, that has previously been saturated with iodine vapour by placing iodine R in an evaporating dish at the bottom of the chamber. After some time brown or yellowish-brown spots become visible. Remove the plate and allow to stand for a few minutes. When the brown background colour has disappeared, spray with starch solution R. Blue spots appear which may become brown on drying and again become blue after spraying with water R. The chromatogram obtained with the test solution always shows a spot with an RF of about 0.5 (oleic acid) and a spot with an RF of about 0.65 (linoleic acid) corresponding to the spots in the chromatogram obtained with the reference solution. With some oils a spot with an RF of about 0.75 may be present (linolenic acid). By comparison with the spot in the chromatogram obtained with the reference solution, verify the absence in the chromatogram obtained with the test solution of a spot with an RF of about 0.25 (erucic acid).
Test for Foreign Oils by Gas Chromatography
The test for foreign oils is carried out on the methyl esters of the fatty acids contained in the oil to be examined by gas chromatography (2.2.28).
METHOD A
This method is not applicable to oils that contain glycerides of fatty acids with an epoxy-, hydroepoxy-, hydroperoxy-, cyclopropyl or cyclopropenyl group, or those that contain a large proportion of fatty acids of chain length less than 8 carbon atoms or to oils with an acid value greater than 2.0.
Test solution When prescribed in the monograph, dry the oil to be examined before the methylation step. Weigh 1.0 g of the oil into a 25 mL round-bottomed flask with a ground-glass neck fitted with a reflux condenser and a gas port into the flask. Add 10 mL of anhydrous methanol R and 0.2 mL of a 60 g/L solution of potassium hydroxide R in methanol R. Attach the reflux condenser, pass nitrogen R through the mixture at a rate of about 50 mL/min, shake and heat to boiling. When the solution is clear (usually after about 10 min), continue heating for a further 5 min. Cool the flask under running water and transfer the contents to a separating funnel. Rinse the flask with 5 mL of heptane R and transfer the rinsings to the separating funnel and shake. Add 10 mL of a 200 g/L solution of sodium chloride R and shake vigorously. Allow to separate and transfer the organic layer to a vial containing anhydrous sodium sulfate R. Allow to stand, then filter.
Reference solution (a) Prepare 0.50 g of the mixture of calibrating substances with the composition described in one of the 2.4.22 tables, as prescribed in the individual monograph (if the monograph does not mention a specific solution, use the composition described in Table 2.4.22.-1). Dissolve in heptane R and dilute to 50.0 mL with the same solvent.
Reference solution (b) Dilute 1.0 mL of reference solution (a) to 10.0 mL with heptane R.
Reference solution (c) Prepare 0.50 g of a mixture of fatty acid methyl esters that corresponds in composition to the mixture of fatty acids indicated in the monograph of the substance to be examined. Dissolve in heptane R and dilute to 50.0 mL with the same solvent. Commercially available mixtures of fatty acid methyl esters may also be used.
Carrier gas helium for chromatography R or hydrogen for chromatography R.
Flow rate 1.3 mL/min (for a column Ø = 0.32 mm).
Split ratio 1:100 or less, according to the internal diameter of the column used (1:50 when Ø = 0.32 mm).
Detection Flame ionisation.
Injection 1 µL.
System suitability When using the mixture of calibrating substances in Table 2.4.22.-1 or Table 2.4.22.-3:
System suitability When using the mixture of calibrating substances in Table 2.4.22.-2:
Assessment of chromatograms
Avoid working conditions tending to give masked peaks (presence of constituents with small differences between retention times, for example linolenic acid and arachidic acid).
Qualitative analysis
Identify the peaks in the chromatogram obtained with reference solution (c) (isothermal operating conditions or linear temperature programming).
When using isothermal operating conditions, the peaks may also be identified by drawing calibration curves using the chromatogram obtained with reference solution (a) and the information given in Tables 2.4.22.-1, 2.4.22.-2 or 2.4.22.-3.



Measure the reduced retention time (t′R) of each peak in the chromatogram obtained with reference solution (a). t′R is the retention time measured from the solvent peak and not from the time of injection. Plot the straight line:

The logarithms of t′R of unsaturated acids are situated on this line at points corresponding to non-integer values of carbon atoms known as ‘equivalent chain lengths′; the equivalent chain length is the length of the theoretical saturated chain that would have the same t′R as the fatty acid to be identified. For example, linoleic acid has the same t′R as the theoretical saturated fatty acid having 18.8 carbon atoms.
Identify the peaks in the chromatogram obtained with the test solution by means of the straight line and the reduced retention times. Equivalent chain lengths are given in Table 2.4.22.-4.

Quantitative analysis
In general, the normalisation procedure is used in which the sum of the areas of the peaks in the chromatogram, except that of the solvent, is set at 100 per cent. The content of a constituent is calculated by determining the area of the corresponding peak as a percentage of the sum of the areas of all the peaks. Disregard any peak with an area less than 0.05 per cent of the total area.
In certain cases, for example in the presence of fatty acids with 12 or less carbon atoms, correction factors can be prescribed in the individual monograph to convert peak areas in per cent m/m.
METHOD B
This method is not applicable to oils that contain glycerides of fatty acids with an epoxy-, hydroepoxy-, hydroperoxy-, cyclopropyl or cyclopropenyl group or to oils with an acid value greater than 2.0.
Test solution Introduce 0.100 g of the substance to be examined into a 10 mL centrifuge tube with a screw cap. Dissolve with 1 mL of heptane R and 1 mL of dimethyl carbonate R and mix vigorously under gentle heating (50-60 °C). Add, while still warm, 1 mL of a 12 g/L solution of sodium R in anhydrous methanol R, prepared with the necessary precautions, and mix vigorously for about 5 min. Add 3 mL of distilled water R and mix vigorously for about 30 s. Centrifuge for 15 min at 1500 g. Inject 1 µL of the organic phase.
Reference solutions and assessment of chromatograms Where there is no specific prescription in the individual monograph, proceed as described under Method A.
Carrier gas helium for chromatography R.
Flow rate 0.9 mL/min.
Split ratio 1:100.

Detection Flame ionisation.
Injection 1 µL.
METHOD C
This method is not applicable to oils that contain glycerides of fatty acids with epoxy-, hydroepoxy-, hydroperoxy-, aldehyde, ketone, cyclopropyl and cyclopropenyl groups, and conjugated polyunsaturated and acetylenic compounds because of partial or complete destruction of these groups.
Test solution Dissolve 0.10 g of the substance to be examined in 2 mL of a 20 g/L solution of sodium hydroxide R in methanol R in a 25 mL conical flask and boil under a reflux condenser for 30 min. Add 2.0 mL of boron trifluoride-methanol solution R through the condenser and boil for 30 min. Add 4 mL of heptane R through the condenser and boil for 5 min. Cool and add 10.0 mL of saturated sodium chloride solution R, shake for about 15 s and add a quantity of saturated sodium chloride solution R such that the upper phase is brought into the neck of the flask. Collect 2 mL of the upper phase, wash with 3 quantities, each of 2 mL, of water R and dry over anhydrous sodium sulfate R.
Reference solutions, chromatographic procedure and assessment of chromatograms Where there is no specific prescription in the individual monograph, proceed as described under Method A.