Appendix XII C. Consistency of Formulated Preparations
1. Uniformity of Weight (Mass)
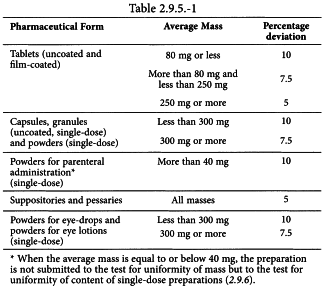
Weigh individually 20 units taken at random or, for single-dose preparations presented in individual containers, the contents of 20 units, and determine the average mass. Not more than 2 of the individual masses deviate from the average mass by more than the percentage deviation shown in Table 2.9.5.-1 and none deviates by more than twice that percentage.
For capsules and powders for parenteral administration, proceed as described below.
CAPSULES
Weigh an intact capsule. Open the capsule without losing any part of the shell and remove the contents as completely as possible. For soft shell capsules, wash the shell with a suitable solvent and allow to stand until the odour of the solvent is no longer perceptible. Weigh the shell. The mass of the contents is the difference between the weighings. Repeat the procedure with another 19 capsules.
POWDERS FOR PARENTERAL ADMINISTRATION
Remove any paper labels from a container and wash and dry the outside. Open the container and without delay weigh the container and its contents. Empty the container as completely as possible by gentle tapping, rinse it if necessary with water R and then with alcohol R and dry at 100-105 °C for 1 h, or, if the nature of the container precludes heating at this temperature, dry at a lower temperature to constant mass. Allow to cool in a desiccator and weigh. The mass of the contents is the difference between the weighings. Repeat the procedure with another 19 containers.
2. Uniformity of Weight (Mass) of Delivered Doses from Multidose Containers
The following test is intended for oral dosage forms such as granules, powders for oral use and liquids for oral use, which are supplied in multidose containers provided at manufacture with a measuring device.
Weigh individually 20 doses taken at random from one or more containers with the measuring device provided and determine the individual and average masses. Not more than 2 of the individual masses deviate from the average mass by more than 10 per cent and none deviates by more than 20 per cent.
3. Uniformity of Content
The test for uniformity of content of single-dose preparations is based on the assay of the individual contents of active substance(s) of a number of single-dose units to determine whether the individual contents are within limits set with reference to the average content of the sample.
The test is not required for multivitamin and trace-element preparations and in other justified and authorised circumstances.
Method Using a suitable analytical method, determine the individual contents of active substance(s) of 10 dosage units taken at random.
Apply the criteria of test A, test B or test C as specified in the monograph for the dosage form in question.
TEST A
The preparation complies with the test if each individual content is between 85 per cent and 115 per cent of the average content. The preparation fails to comply with the test if more than one individual content is outside these limits or if one individual content is outside the limits of 75 per cent to 125 per cent of the average content.
If one individual content is outside the limits of 85 per cent to 115 per cent but within the limits of 75 per cent to 125 per cent, determine the individual contents of another 20 dosage units taken at random. The preparation complies with the test if not more than one of the individual contents of the 30 units is outside 85 per cent to 115 per cent of the average content and none is outside the limits of 75 per cent to 125 per cent of the average content.
TEST B
The preparation complies with the test if not more than one individual content is outside the limits of 85 per cent to 115 per cent of the average content and none is outside the limits of 75 per cent to 125 per cent of the average content. The preparation fails to comply with the test if more than 3 individual contents are outside the limits of 85 per cent to 115 per cent of the average content or if one or more individual contents are outside the limits of 75 per cent to 125 per cent of the average content.
If 2 or 3 individual contents are outside the limits of 85 per cent to 115 per cent but within the limits of 75 per cent to 125 per cent, determine the individual contents of another 20 dosage units taken at random. The preparation complies with the test if not more than 3 individual contents of the 30 units are outside the limits of 85 per cent to 115 per cent of the average content and none is outside the limits of 75 per cent to 125 per cent of the average content.
TEST C
The preparation complies with the test if the average content of the 10 dosage units is between 90 per cent and 110 per cent of the content stated on the label and if the individual content of each dosage unit is between 75 per cent and 125 per cent of the average content.
4. Uniformity of Dosage Units1
To ensure the consistency of dosage units, each unit in a batch should have an active substance content within a narrow range around the label claim. Dosage units are defined as dosage forms containing a single dose or a part of a dose of an active substance in each dosage unit. ♢Unless otherwise stated,♢ the uniformity of dosage units specification is not intended to apply to solutions, suspensions, emulsions or gels in single-dose containers intended for local action following cutaneous administration. ♢The test for content uniformity is not required for multivitamin, single-vitamin and trace-element preparations.♢
The term ‘uniformity of dosage unit’ is defined as the degree of uniformity in the amount of the active substance among dosage units. Therefore, the requirements of this chapter apply to each active substance being comprised in dosage units containing 1 or more active substances, unless otherwise specified elsewere in this Pharmacopoeia.
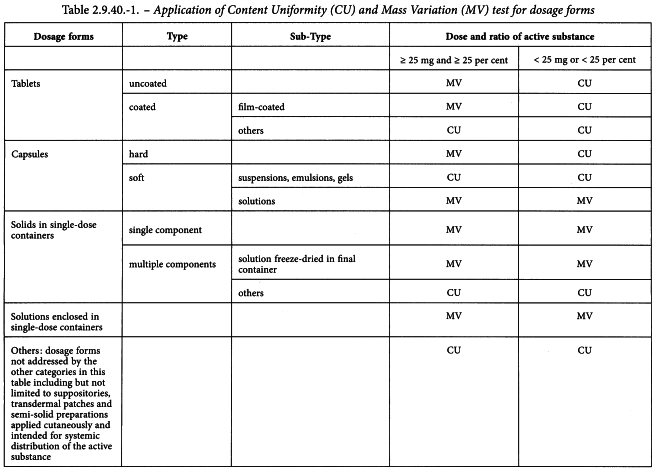
The uniformity of dosage units can be demonstrated by either of 2 methods: content uniformity or mass variation (see Table 2.9.40.-1).
The test for content uniformity of preparations presented in dosage units is based on the assay of the individual contents of active substance(s) of a number of dosage units to determine whether the individual contents are within the limits set. The content uniformity method may be applied in all cases.
The test for mass variation is applicable for the following dosage forms:
(1) solutions enclosed in single-dose containers and in soft capsules;
(2) solids (including powders, granules and sterile solids) that are packaged in single-dose containers and contain no added active or inactive substances;
(3) solids (including sterile solids) that are packaged in single-dose containers, with or without added active or inactive substances, that have been prepared from true solutions and freeze-dried in the final containers and are labelled to indicate this method of preparation;
(4) hard capsules, uncoated tablets, or film-coated tablets, containing 25 mg or more of an active substance comprising 25 per cent or more, by mass, of the dosage unit or, in the case of hard capsules, the capsule contents, except that uniformity of other active substances present in lesser proportions is demonstrated by meeting content uniformity requirements.
The test for content uniformity is required for all dosage forms not meeting the above conditions for the mass variation test. ♦Alternatively, products that do not meet the 25 mg/25 per cent threshold limit may be tested for uniformity of dosage units by mass variation instead of the content uniformity test on the following condition: the concentration Relative Standard Deviation (RSD) of the active substance in the final dosage units is not more than 2 per cent, based on process validation data and development data, and if there has been regulatory approval of such a change. The concentration RSD is the RSD of the concentration per dosage unit (m/m or m/V), where concentration per dosage unit equals the assay result per dosage unit divided by the individual dosage unit mass. See the RSD formula in Table 2.9.40.-2.♦
CONTENT UNIFORMITY
Select not fewer than 30 units, and proceed as follows for the dosage form designated. Where different procedures are used for assay of the preparation and for the content uniformity test, it may be necessary to establish a correction factor to be applied to the results of the latter.
Solid dosage forms
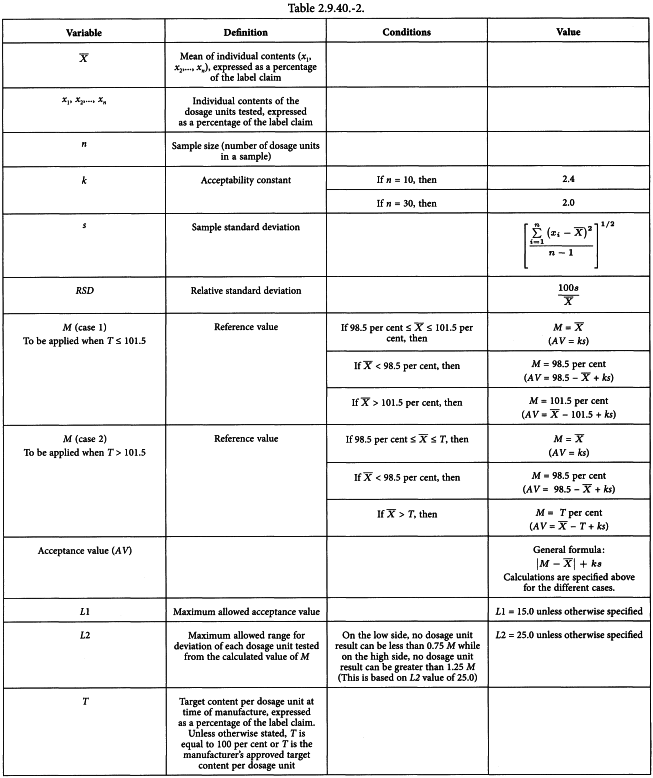
Assay 10 units individually using an appropriate analytical method. Calculate the acceptance value (see Table 2.9.40.-2).
Liquid or semi-solid dosage forms
Assay 10 units individually using an appropriate analytical method. Carry out the assay on the amount of well-mixed material that is removed from an individual container in conditions of normal use. Express the results as delivered dose. Calculate the acceptance value (see Table 2.9.40.-2).
Calculation of Acceptance Value
Calculate the Acceptance Value (AV) using the formula:

for which the terms are as defined in Table 2.9.40.-2.
MASS VARIATION
Carry out an assay for the active substance(s) on a representative sample of the batch using an appropriate analytical method. This value is result A, expressed as percentage of label claim (see Calculation of Acceptance Value). Assume that the concentration (mass of active substance per mass of dosage unit) is uniform. Select not fewer than 30 dosage units, and proceed as follows for the dosage form designated.
Uncoated or film-coated tablets
Accurately weigh 10 tablets individually. Calculate the active substance content, expressed as percentage of label claim, of each tablet from the mass of the individual tablets and the result of the assay. Calculate the acceptance value.
Hard capsules
Accurately weigh 10 capsules individually, taking care to preserve the identity of each capsule. Remove the contents of each capsule by suitable means. Accurately weigh the emptied shells individually, and calculate for each capsule the net mass of its contents by subtracting the mass of the shell from the respective gross mass. Calculate the active substance content in each capsule from the mass of product removed from the individual capsules and the result of the assay. Calculate the acceptance value.
Soft capsules
Accurately weigh 10 intact capsules individually to obtain their gross masses, taking care to preserve the identity of each capsule. Then cut open the capsules by means of a suitable clean, dry cutting instrument such as scissors or a sharp open blade, and remove the contents by washing with a suitable solvent. Allow the occluded solvent to evaporate from the shells at room temperature over a period of about 30 min, taking precautions to avoid uptake or loss of moisture. Weigh the individual shells, and calculate the net contents. Calculate the active substance content in each capsule from the mass of product removed from the individual capsules and the result of the assay. Calculate the acceptance value.
Solid dosage forms other than tablets and capsules
Proceed as directed for hard capsules, treating each unit as described therein. Calculate the acceptance value.
Liquid ♢or semi-solid♢ dosage forms
Accurately weigh the amount of liquid or semi-solid that is removed from each of 10 individual containers in conditions of normal use. If necessary, compute the equivalent volume after determining the density. Calculate the active substance content in each container from the mass of product removed from the individual containers and the result of the assay. Calculate the acceptance value.
Calculation of Acceptance Value
Calculate the acceptance value (AV) as shown in content uniformity, except that the individual contents of the units are replaced with the individual estimated contents defined below.
x1, x2,..., xn | = | individual estimated contents of the dosage units tested; |
where

w1, w2,..., wn | = | individual masses of the dosage units tested; |
A | = | content of active substance (percentage of label claim) obtained using an appropriate analytical method (assay); |
 | = | mean of individual masses (w1, w2,..., wn). |
CRITERIA
Apply the following criteria, unless otherwise specified.
Solid, semi-solid and liquid dosage forms
The requirements for dosage uniformity are met if the acceptance value of the first 10 dosage units is less than or equal to L1 per cent. If the acceptance value is greater than L1 per cent, test the next 20 dosage units and calculate the acceptance value. The requirements are met if the final acceptance value of the 30 dosage units is less than or equal to L1 per cent and no individual content of the dosage unit is less than (1 -L2 × 0.01)M or more than (1 + L2 × 0.01)M in calculation of acceptance value under content uniformity or under mass variation. Unless otherwise specified, L1 is 15.0 and L2 is 25.0.
5. Extractable Volume of Parenteral Preparations1
Suspensions and emulsions are shaken before withdrawal of the contents and before the determination of the density. Oily and viscous preparations may be warmed according to the instructions on the label, if necessary, and thoroughly shaken immediately before removing the contents. The contents are then cooled to 20-25 °C before measuring the volume.
SINGLE-DOSE CONTAINERS
Select 1 container if the nominal volume is 10 mL or more, 3 containers if the nominal volume is more than 3 mL and less than 10 mL, or 5 containers if the nominal volume is 3 mL or less. Take up individually the total contents of each container selected into a dry syringe of a capacity not exceeding 3 times the volume to be measured, and fitted with a 21-gauge needle not less than 2.5 cm in length. Expel any air bubbles from the syringe and needle, then discharge the contents of the syringe without emptying the needle into a standardised dry cylinder (graduated to contain rather than to deliver the designated volumes) of such size that the volume to be measured occupies at least 40 per cent of its graduated volume. Alternatively, the volume of the contents in millilitres may be calculated as the mass in grams divided by the density.
For containers with a nominal volume of 2 mL or less, the contents of a sufficient number of containers may be pooled to obtain the volume required for the measurement provided that a separate, dry syringe assembly is used for each container. The contents of containers holding 10 mL or more may be determined by opening them and emptying the contents directly into the graduated cylinder or tared beaker.
The volume is not less than the nominal volume in case of containers examined individually, or, in case of containers with a nominal volume of 2 mL or less, is not less than the sum of the nominal volumes of the containers taken collectively.
MULTIDOSE CONTAINERS
For injections in multidose containers labelled to yield a specific number of doses of a stated volume, select one container and proceed as directed for single-dose containers using the same number of separate syringe assemblies as the number of doses specified.
The volume is such that each syringe delivers not less than the stated dose.
CARTRIDGES AND PREFILLED SYRINGES
Select 1 container if the nominal volume is 10 mL or more, 3 containers if the nominal volume is more than 3 mL and less than 10 mL, or 5 containers if the nominal volume is 3 mL or less. If necessary, fit the containers with the accessories required for their use (needle, piston, syringe) and transfer the entire contents of each container without emptying the needle into a dry tared beaker by slowly and constantly depressing the piston. Determine the volume in millilitres calculated as the mass in grams divided by the density.
The volume measured for each of the containers is not less than the nominal volume.
PARENTERAL INFUSIONS
Select one container. Transfer the contents into a dry measuring cylinder of such a capacity that the volume to be determined occupies at least 40 per cent of the nominal volume of the cylinder. Measure the volume transferred.
The volume is not less than the nominal volume.
7. Preparations for Inhalation: Aerodynamic Assessment of Fine Particles
This test is used to determine the fine particle characteristics of the aerosol clouds generated by preparations for inhalation.
Unless otherwise justified and authorised, one of the following apparatus and test procedures is used.
Stage mensuration Is performed periodically together with confirmation of other dimensions critical to the effective operation of the impactor.
Re-entrainment (for apparatus D and E) To ensure efficient particle capture, coat each plate with glycerol, silicone oil or similar high viscosity liquid, typically deposited from a volatile solvent. Plate coating must be part of method validation and may be omitted where justified and authorised.
Mass balance The total mass of the active substance is not less than 75 per cent and not more than 125 per cent of the average delivered dose determined during testing for uniformity of delivered dose. This is not a test of the inhaler but it serves to ensure that the results are valid.
APPARATUS A - GLASS IMPINGER
The apparatus is shown in Figure 2.9.18.-1 (see also Table 2.9.18.-1).
Procedure for nebulisers
Introduce 7 mL and 30 mL of a suitable solvent into the upper and lower impingement chambers, respectively.
Connect all the component parts. Ensure that the assembly is vertical and adequately supported and that the jet spacer peg of the lower jet assembly just touches the bottom of the lower impingement chamber. Connect a suitable pump fitted with a filter (of suitable pore size) to the outlet of the apparatus. Adjust the air flow through the apparatus, as measured at the inlet to the throat, to 60 ± 5 L/min.
Introduce the liquid preparation for inhalation into the reservoir of the nebuliser. Fit the mouthpiece and connect it by means of an adapter to the device.
Switch on the pump of the apparatus and after 10 s switch on the nebuliser.
After 60 s, unless otherwise justified, switch off the nebuliser, wait for about 5 s and then switch off the pump of the apparatus. Dismantle the apparatus and wash the inner surface of the upper impingement chamber collecting the washings in a volumetric flask. Wash the inner surface of the lower impingement chamber collecting the washings in a second volumetric flask. Finally, wash the filter preceding the pump and its connections to the lower impingement chamber and combine the washings with those obtained from the lower impingement chamber. Determine the amount of active substance collected in each of the 2 flasks. Express the results for each of the 2 parts of the apparatus as a percentage of the total amount of active substance.
Procedure for pressurised inhalers
Place the actuator adapter in position at the end of the throat so that the mouthpiece end of the actuator, when inserted to a depth of about 10 mm, lines up along the horizontal axis of the throat and the open end of the actuator, which accepts the pressurised container, is uppermost and in the same vertical plane as the rest of the apparatus.
Introduce 7 mL and 30 mL of a suitable solvent into the upper and lower impingement chambers, respectively.
Connect all the component parts. Ensure that the assembly is vertical and adequately supported and that the lower jet-spacer peg of the lower jet assembly just touches the bottom of the lower impingement chamber. Connect a suitable pump to the outlet of the apparatus. Adjust the air flow through the apparatus, as measured at the inlet to the throat, to 60 ± 5 L/min.
Prime the metering valve by shaking for 5 s and discharging once to waste; after not less than 5 s, shake and discharge again to waste. Repeat a further 3 times.
Shake for about 5 s, switch on the pump to the apparatus and locate the mouthpiece end of the actuator in the adapter, discharge once immediately. Remove the assembled inhaler from the adapter, shake for not less than 5 s, relocate the mouthpiece end of the actuator in the adapter and discharge again. Repeat the discharge sequence. The number of discharges should be minimised and typically would not be greater than 10. After the final discharge wait for not less than 5 s and then switch off the pump. Dismantle the apparatus.
Wash the inner surface of the inlet tube to the lower impingement chamber and its outer surface that projects into the chamber with a suitable solvent, collecting the washings in the lower impingement chamber. Determine the content of active substance in this solution. Calculate the amount of active substance collected in the lower impingement chamber per discharge and express the results as a percentage of the dose stated on the label.
Procedure for powder inhalers
Introduce 7 mL and 30 mL of a suitable solvent into the upper and lower impingement chambers, respectively.
Connect all the component parts. Ensure that the assembly is vertical and adequately supported and that the jet-spacer peg of the lower jet assembly just touches the bottom of the lower impingement chamber. Without the inhaler in place, connect a suitable pump to the outlet of the apparatus. Adjust the air flow through the apparatus, as measured at the inlet to the throat, to 60 ± 5 L/min.
Prepare the inhaler for use and locate the mouthpiece in the apparatus by means of a suitable adapter. Switch on the pump for 5 s. Switch off the pump and remove the inhaler. Repeat the discharge sequence. The number of discharges should be minimised and typically would not be greater than 10. Dismantle the apparatus.
Wash the inner surface of the inlet tube to the lower impingement chamber and its outer surface that projects into the chamber with a suitable solvent, collecting the washings in the lower impingement chamber. Determine the content of active substance in this solution. Calculate the amount of active substance collected in the lower impingement chamber per discharge and express the results as a percentage of the dose stated on the label.


Fine particle dose and particle size distribution
APPARATUS C - MULTI-STAGE LIQUID IMPINGER
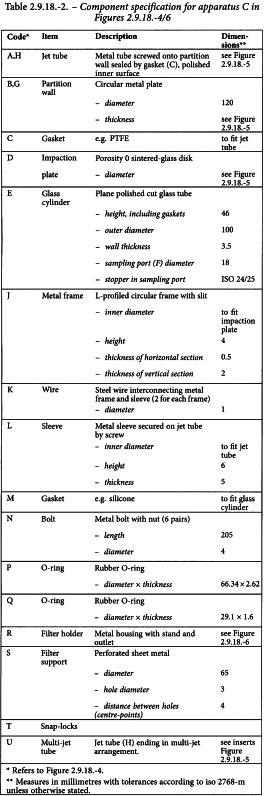
The multi-stage liquid impinger consists of impaction stages 1 (pre-separator), 2, 3 and 4 and an integral filter stage (stage 5), see Figures 2.9.18.-4/6. An impaction stage comprises an upper horizontal metal partition wall (B) through which a metal inlet jet tube (A) with its impaction plate (D) is protruding. A glass cylinder (E) with sampling port (F) forms the vertical wall of the stage, and a lower horizontal metal partition wall (G) through which the tube (H) connects to the next lower stage. The tube into stage 4 (U) ends in a multi-jet arrangement. The impaction plate (D) is secured in a metal frame (J) which is fastened by 2 wires (K) to a sleeve (L) secured on the jet tube. The horizontal face of the collection plate is perpendicular to the axis of the jet tube and centrally aligned. The upper surface of the impaction plate is slightly raised above the edge of the metal frame. A recess around the perimeter of the horizontal partition wall guides the position of the glass cylinder. The glass cylinders are sealed against the horizontal partition walls with gaskets (M) and clamped together by 6 bolts (N). The sampling ports are sealed by stoppers. The bottom-side of the lower partition wall of stage 4 has a concentrical protrusion fitted with a rubber O-ring (P) which seals against the edge of a filter placed in the filter holder. The filter holder (R) is constructed as a basin with a concentrical recess in which a perforated filter support (S) is flush-fitted. The filter holder is dimensioned for 76 mm diameter filters. The assembly of impaction stages is clamped onto the filter holder by 2 snap-locks (T). Connect an induction port (see Figure 2.9.18.-7) onto the stage 1 inlet jet tube of the impinger. A rubber O-ring on the jet tube provides an airtight connection to the induction port. A suitable mouthpiece adapter is used to provide an airtight seal between the inhaler and the induction port. The front face of the inhaler mouthpiece must be flush with the front face of the induction port.


Procedure for pressurised inhalers
Dispense 20 mL of a solvent, capable of dissolving the active substance into each of stages 1 to 4 and replace the stoppers. Tilt the apparatus to wet the stoppers, thereby neutralising electrostatic charge. Place a suitable filter capable of quantitatively collecting the active substance in stage 5 and assemble the apparatus. Place a suitable mouthpiece adapter in position at the end of the induction port so that the mouthpiece end of the actuator, when inserted, lines up along the horizontal axis of the induction port and the inhaler is positioned in the same orientation as intended for use. Connect a suitable vacuum pump to the outlet of the apparatus and adjust the air flow through the apparatus, as measured at the inlet to the induction port, to 30 L/min (± 5 per cent). Switch off the pump.
Unless otherwise prescribed in the patient instructions, shake the inhaler for 5 s and discharge 1 delivery to waste. Switch on the pump to the apparatus, locate the mouthpiece end of the actuator in the adapter and discharge the inhaler into the apparatus, depressing the valve for a sufficient time to ensure complete discharge. Wait for 5 s before removing the assembled inhaler from the adapter. Repeat the procedure. The number of discharges should be minimised and typically would not be greater than 10. The number of discharges is sufficient to ensure an accurate and precise determination of the fine particle dose. After the final discharge, wait for 5 s and then switch off the pump.
Dismantle the filter stage of the apparatus. Carefully remove the filter and extract the active substance into an aliquot of the solvent. Remove the induction port and mouthpiece adapter from the apparatus and extract the active substance into an aliquot of the solvent. If necessary, rinse the inside of the inlet jet tube to stage 1 with solvent, allowing the solvent to flow into the stage. Extract the active substance from the inner walls and the collection plate of each of the 4 upper stages of the apparatus into the solution in the respective stage by carefully tilting and rotating the apparatus, observing that no liquid transfer occurs between the stages.
Using a suitable method of analysis, determine the quantity of active substance contained in each of the aliquots of solvent.
Calculate the fine particle dose (see Calculations).



1. Material may be aluminium, stainless steel or other suitable material. | |
2. Machine from 38 mm bar stock. | |
3. Bore 19 mm hole through bar. | |
4. Cut tube to exact 45° as shown. | |
5. The inner bores and tapers should be smooth – surface roughness Ra approx. 0.4 µm. | |
6. Mill joining cads of stock to provide a liquid tight leak-free seal. | |
7. Set up a holding fixture for aligning the inner 19 mm bore and for drilling and tapping M4 × 0.7 threads. There must be virtually no mismatch of the inner bores in the miter joint. |
Procedure for powder inhalers
Place a suitable low resistance filter capable of quantitatively collecting the active substance in stage 5 and assemble the apparatus. Connect the apparatus to a flow system according to the scheme specified in Figure 2.9.18.-8 and Table 2.9.18.-4. Unless otherwise defined, conduct the test at the flow rate, Qout, used in the test for uniformity of delivered dose, drawing 4 L of air from the mouthpiece of the inhaler and through the apparatus.
Connect a flowmeter to the induction port. Use a flowmeter calibrated for the volumetric flow leaving the meter, or calculate the volumetric flow leaving the meter (Qout) using the ideal gas law. For a meter calibrated for the entering volumetric flow (Qin), use the following expression:

P0 | = | atmospheric pressure, |
ΔP | = | pressure drop over the meter. |
Adjust the flow control valve to achieve steady flow through the system at the required rate, Qout (± 5 per cent). Switch off the pump. Ensure that critical flow occurs in the flow control valve by the following procedure.
With the inhaler in place and the test flow rate established, measure the absolute pressure on both sides of the control valve (pressure reading points P2 and P3 in Figure 2.9.18.-8). A ratio P3/P2 of less than or equal to 0.5 indicates critical flow. Switch to a more powerful pump and re-measure the test flow rate if critical flow is not indicated.
Dispense 20 mL of a solvent, capable of dissolving the active substance into each of the 4 upper stages of the apparatus and replace the stoppers. Tilt the apparatus to wet the stoppers, thereby neutralising electrostatic charge. Place a suitable mouthpiece adapter in position at the end of the induction port.


Prepare the powder inhaler for use according to patient instructions. With the pump running and the 2-way solenoid valve closed, locate the mouthpiece of the inhaler in the mouthpiece adapter. Discharge the powder into the apparatus by opening the valve for the required time, T (± 5 per cent). Repeat the procedure. The number of discharges should be minimised and typically would not be greater than 10. The number of discharges is sufficient to ensure an accurate and precise determination of fine particle dose.
Dismantle the filter stage of the apparatus. Carefully remove the filter and extract the active substance into an aliquot of the solvent. Remove the induction port and mouthpiece adapter from the apparatus and extract the active substance into an aliquot of the solvent. If necessary, rinse the inside of the inlet jet tube to stage 1 with solvent, allowing the solvent to flow into the stage. Extract the active substance from the inner walls and the collection plate of each of the 4 upper stages of the apparatus into the solution in the respective stage by carefully tilting and rotating the apparatus, observing that no liquid transfer occurs between the stages.
Using a suitable method of analysis, determine the amount of active substance contained in each of the aliquots of solvent.
Calculate the fine particle dose (see Calculations).
APPARATUS D - ANDERSEN cascade impactor
The Andersen 1 ACFM non-viable cascade impactor consists of 8 stages together with a final filter. Material of construction may be aluminium, stainless steel or other suitable material. The stages are clamped together and sealed with O-rings. Critical dimensions applied by the manufacturer of apparatus D are provided in Table 2.9.18.-5. In use, some occlusion and wear of holes will occur. In-use mensuration tolerances need to be justified. In the configuration used for pressurised inhalers (Figure 2.9.18.-9) the entry cone of the impactor is connected to an induction port (see Figure 2.9.18.-7). A suitable mouthpiece adapter is used to provide an airtight seal between the inhaler and the induction port. The front face of the inhaler mouthpiece must be flush with the front face of the induction port.

In the configuration for powder inhalers, a pre-separator is placed above the top stage to collect large masses of non-respirable powder. It is connected to the induction port as shown in Figure 2.9.18.-10. To accommodate high flow rates through the impactor, the outlet nipple, used to connect the impactor to the vacuum system is enlarged to have an internal diameter of greater than or equal to 8 mm.


Procedure for pressurised inhalers
Assemble the Andersen impactor with a suitable filter in place. Ensure that the system is airtight. In that respect, follow the manufacturer′s instructions. Place a suitable mouthpiece adapter in position at the end of the induction port so that the mouthpiece end of the actuator, when inserted, lines up along the horizontal axis of the induction port and the inhaler unit is positioned in the same orientation as the intended use. Connect a suitable pump to the outlet of the apparatus and adjust the air flow through the apparatus, as measured at the inlet to the induction port, to 28.3 L/min (± 5 per cent). Switch off the pump.
Unless otherwise prescribed in the patient instructions, shake the inhaler for 5 s and discharge one delivery to waste. Switch on the pump to the apparatus, locate the mouthpiece end of the actuator in the adapter and discharge the inverted inhaler into the apparatus, depressing the valve for a sufficient time to ensure complete discharge. Wait for 5 s before removing the assembled inhaler from the adapter. Repeat the procedure. The number of discharges should be minimised and typically would not be greater than 10. The number of discharges is sufficient to ensure an accurate and precise determination of the fine particle dose. After the final discharge, wait for 5 s and then switch off the pump.
Dismantle the apparatus. Carefully remove the filter and extract the active substance into an aliquot of the solvent. Remove the induction port and mouthpiece adapter from the apparatus and extract the active substance into an aliquot of the solvent. Extract the active substance from the inner walls and the collection plate of each of the stages of the apparatus into aliquots of solvent.
Using a suitable method of analysis, determine the quantity of active substance contained in each of the aliquots of solvent.
Calculate the fine particle dose (see Calculations).
Procedure for powder inhalers
The aerodynamic cut-off diameters of the individual stages of this apparatus are currently not well-established at flow rates other than 28.3 L/min. Users must justify and validate the use of the impactor in the chosen conditions, when flow rates different from 28.3 L/min are selected.
Assemble the Andersen impactor with the pre-separator and a suitable filter in place and ensure that the system is airtight. Depending on the product characteristics, the pre-separator may be omitted, where justified and authorised. Stages 6 and 7 may also be omitted at high flow rates, if justified. The pre-separator may be coated in the same way as the plates or may contain 10 mL of a suitable solvent. Connect the apparatus to a flow system according to the scheme specified in Figure 2.9.18.-8 and Table 2.9.18.-4.
Unless otherwise defined, conduct the test at the flow rate, Qout, used in the test for uniformity of delivered dose drawing 4 L of air from the mouthpiece of the inhaler and through the apparatus.
Connect a flowmeter to the induction port. Use a flowmeter calibrated for the volumetric flow leaving the meter, or calculate the volumetric flow leaving the meter (Qout) using the ideal gas law. For a meter calibrated for the entering volumetric flow (Qin), use the following expression:

P0 | = | atmospheric pressure, |
ΔP | = | pressure drop over the meter. |
Adjust the flow control valve to achieve steady flow through the system at the required rate, Qout (± 5 per cent). Ensure that critical flow occurs in the flow control valve by the procedure described for Apparatus C. Switch off the pump.
Prepare the powder inhaler for use according to the patient instructions. With the pump running and the 2-way solenoid valve closed, locate the mouthpiece of the inhaler in the mouthpiece adapter. Discharge the powder into the apparatus by opening the valve for the required time, T (± 5 per cent). Repeat the discharge sequence. The number of discharges should be minimised and typically would not be greater than 10. The number of discharges is sufficient to ensure an accurate and precise determination of fine particle dose.
Dismantle the apparatus. Carefully remove the filter and extract the active substance into an aliquot of the solvent. Remove the pre-separator, induction port and mouthpiece adapter from the apparatus and extract the active substance into an aliquot of the solvent. Extract the active substance from the inner walls and the collection plate of each of the stages of the apparatus into aliquots of solvent.
Using a suitable method of analysis, determine the quantity of active substance contained in each of the aliquots of solvent.
Calculate the fine particle dose (see Calculations).
APPARATUS E
Apparatus E is a cascade impactor with 7 stages and a micro-orifice collector (MOC). Over the flow rate range of 30 L/min to 100 L/min the 50 per cent-efficiency cut-off diameters (D50 values) range between 0.24 µm to 11.7 µm, evenly spaced on a logarithmic scale. In this flow range, there are always at least 5 stages with D50 values between 0.5 µm and 6.5 µm. The collection efficiency curves for each stage are sharp and minimise overlap between stages.
Material of construction may be aluminium, stainless steel or other suitable material.
The impactor configuration has removable impaction cups with all the cups in one plane (Figures 2.9.18.-11/14). There are 3 main sections to the impactor; the bottom frame that holds the impaction cups, the seal body that holds the jets and the lid that contains the interstage passageways (Figures 2.9.18.-11/12). Multiple nozzles are used at all but the first stage (Figure 2.9.18.-13). The flow passes through the impactor in a saw-tooth pattern.
Critical dimensions are provided in Table 2.9.18.-6.

In routine operation, the seal body and lid are held together as a single assembly. The impaction cups are accessible when this assembly is opened at the end of an inhaler test. The cups are held in a support tray, so that all cups can be removed from the impactor simultaneously by lifting out the tray.
An induction port with internal dimensions (relevant to the airflow path) defined in Figure 2.9.18.-7 connects to the impactor inlet. A pre-separator can be added when required, typically with powder inhalers, and connects between the induction port and the impactor. A suitable mouthpiece adapter is used to provide an airtight seal between the inhaler and the induction port.
Apparatus E contains a terminal Micro-Orifice Collector (MOC) that for most formulations will eliminate the need for a final filter as determined by method validation. The MOC is an impactor plate with nominally 4032 holes, each approximately 70 µm in diameter. Most particles not captured on stage 7 of the impactor will be captured on the cup surface below the MOC. For impactors operated at 60 L/min, the MOC is capable of collecting 80 per cent of 0.14 µm particles. For formulations with a significant fraction of particles not captured by the MOC, there is an optional filter holder that can replace the MOC or be placed downstream of the MOC (a glass fibre filter is suitable).



Procedure for pressurised inhalers
Place cups into the apertures in the cup tray. Insert the cup tray into the bottom frame, and lower into place. Close the impactor lid with the seal body attached and operate the handle to lock the impactor together so that the system is airtight.
Connect an induction port with internal dimensions defined in Figure 2.9.18.-7 to the impactor inlet. Place a suitable mouthpiece adapter in position at the end of the induction port so that the mouthpiece end of the actuator, when inserted, lines up along the horizontal axis of the induction port. The front face of the inhaler mouthpiece must be flush with the front face of the induction port. When attached to the mouthpiece adapter, the inhaler is positioned in the same orientation as intended for use. Connect a suitable pump to the outlet of the apparatus and adjust the air flow through the apparatus, as measured at the inlet to the induction port, to 30 L/min (± 5 per cent). Switch off the pump.
Unless otherwise prescribed in the patient instructions, shake the inhaler for 5 s and discharge 1 delivery to waste. Switch on the pump to the apparatus. Prepare the inhaler for use according to the patient instructions, locate the mouthpiece end of the actuator in the adapter and discharge the inhaler into the apparatus, depressing the valve for a sufficient time to ensure a complete discharge. Wait for 5 s before removing the assembled inhaler from the adapter. Repeat the procedure. The number of discharges should be minimised, and typically would not be greater than 10. The number of discharges is sufficient to ensure an accurate and precise determination of the fine particle dose. After the final discharge, wait for 5 s and then switch off the pump.
Dismantle the apparatus and recover the active substance as follows: remove the induction port and mouthpiece adapter from the apparatus and recover the deposited active substance into an aliquot of solvent. Open the impactor by releasing the handle and lifting the lid. Remove the cup tray, with the collection cups, and recover the active substance in each cup into an aliquot of solvent.
Using a suitable method of analysis, determine the quantity of active substance contained in each of the aliquots of solvent.
Calculate the fine particle dose (see Calculations).

Procedure for powder inhalers

Assemble the apparatus with the pre-separator (Figure 2.9.18.-15). Depending on the product characteristics, the pre-separator may be omitted, where justified.
Place cups into the apertures in the cup tray. Insert the cup tray into the bottom frame, and lower into place. Close the impactor lid with the seal body attached and operate the handle to lock the impactor together so that the system is airtight.
When used, the pre-separator should be assembled as follows: assemble the pre-separator insert into the pre-separator base. Fit the pre-separator base to the impactor inlet. Add 15 mL of the solvent used for sample recovery to the central cup of the pre-separator insert. Place the pre-separator body on top of this assembly and close the 2 catches.
Connect an induction port with internal dimensions defined in Figure 2.9.18.-7 to the impactor inlet or pre-separator inlet. Place a suitable mouthpiece adapter in position at the end of the induction port so that the mouthpiece end of the inhaler, when inserted, lines up along the horizontal axis of the induction port. The front face of the inhaler mouthpiece must be flush with the front face of the induction port. When attached to the mouthpiece adapter, the inhaler is positioned in the same orientation as intended for use. Connect the apparatus to a flow system according to the scheme specified in Figure 2.9.18.-8 and Table 2.9.18.-4.
Unless otherwise prescribed, conduct the test at the flow rate, Qout, used in the test for uniformity of delivered dose drawing 4 L of air from the mouthpiece of the inhaler and through the apparatus. Connect a flowmeter to the induction port. Use a flowmeter calibrated for the volumetric flow leaving the meter, or calculate the volumetric flow leaving the meter (Qout) using the ideal gas law. For a meter calibrated for the entering volumetric flow (Qin), use the following expression:

P0 | = | atmospheric pressure, |
ΔP | = | pressure drop over the meter. |
Adjust the flow control valve to achieve steady flow through the system at the required rate, Qout (± 5 per cent). Ensure that critical flow occurs in the flow control valve by the procedure described for Apparatus C. Switch off the pump.
Prepare the powder inhaler for use according to the patient instructions. With the pump running and the 2-way solenoid valve closed, locate the mouthpiece of the inhaler in the mouthpiece adapter. Discharge the powder into the apparatus by opening the valve for the required time, T (± 5 per cent). Repeat the discharge sequence. The number of discharges should be minimised and typically would not be greater than 10. The number of discharges is sufficient to ensure an accurate and precise determination of fine particle dose.
Dismantle the apparatus and recover the active substance as follows: remove the induction port and mouthpiece adapter from the pre-separator, when used, and recover the deposited active substance into an aliquot of solvent. When used, remove the pre-separator from the impactor, being careful to avoid spilling the cup liquid into the impactor. Recover the active substance from the pre-separator.
Open the impactor by releasing the handle and lifting the lid. Remove the cup tray, with the collection cups, and recover the active substance in each cup into an aliquot of solvent.
Using a suitable method of analysis, determine the quantity of active substance contained in each of the aliquots of solvent.
Calculate the fine particle dose (see Calculations).
Calculations
From the analysis of the solutions, calculate the mass of active substance deposited on each stage per discharge and the mass of active substance per discharge deposited in the induction port, mouthpiece adapter and when used, the pre-separator.
Starting at the final collection site (filter or MOC), derive a table of cumulative mass versus cut-off diameter of the respective stage (see Tables 2.9.18.-7 for Apparatus C, 2.9.18.-8 for Apparatus D, 2.9.18.-9 for Apparatus E). Calculate by interpolation the mass of the active substance less than 5 µm. This is the Fine Particle Dose (FPD).
If necessary, and where appropriate (e.g., where there is a log-normal distribution), plot the cumulative fraction of active substance versus cut-off diameter (see Tables 2.9.18.-7/9) on log probability paper, and use this plot to determine values for the Mass Median Aerodynamic Diameter (MMAD) and Geometric Standard Deviation (GSD) as appropriate. Appropriate computational methods may also be used.



8. Preparations for Nebulisation: Characterisation
Products used for nebulisation and intended for pulmonary delivery are characterised using the following tests:
These tests standardise the approach given to the assessment of the dose that would be delivered to a patient but are not intended to provide assessment of the nebuliser device itself, which is described in the European standard EN 13544-1:2007+A1:2009, Respiratory therapy equipment - Part 1: Nebulizing systems and their components.
The mass- rather than the number-weighted size distribution is more appropriate to evaluate product performance. Indeed, active substance mass as a function of aerodynamic diameter is more indicative of therapeutic effect within the respiratory tract.
ACTIVE SUBSTANCE DELIVERY RATE AND TOTAL ACTIVE SUBSTANCE DELIVERED
These tests are performed to assess the rate of delivery to the patient and the total active substance delivered to the patient, using standardised conditions of volumetric flow rate. It is essential that breath-enhanced and breath-actuated nebulisers be evaluated by a breathing simulator, as the output of these types of device is highly dependent on inhalation flow rate. The methodology below describes the use of a standard breathing pattern defined for adults. Should a particular product for nebulisation only be indicated for paediatric (i.e. neonate, infant or child) use, then paediatric breathing pattern(s) must be used. Breathing patterns are used, rather than continuous flow rates, to provide a more appropriate measure of the mass of active substance that would be delivered to patients.
Active substance delivery rate and total active substance delivered are appropriate characteristics because they allow the mass delivered to be characterised in a standard way regardless of the nebuliser used. Accordingly, the test methodology described below allows that the mass of active substance delivered in the 1st period (typically 1 min) is measured (consequently giving an assessment of active substance delivery rate) as well as capturing the total mass of active substance delivered.
Apparatus
Breathing simulator
A commercially available breathing simulator, which is able to generate the breathing profiles specified in Table 2.9.44.-1, is used for the test. The breathing profile indicated for adults is used unless the medicinal product is specifically intended for use in paediatrics, where alternate patterns should be used, as indicated in Table 2.9.44.-1.

Filter system
A suitably validated low-resistance filter, capable of quantitatively collecting the aerosol and enabling recovery of the active substance with an appropriate solvent, is used for the test. The dead volume of the filter casing does not exceed 10 per cent of the tidal volume used in the breath simulation.
Method
Attach the filter (contained in the filter holder) (A) to the breath simulator (B) according to Figure 2.9.44.-1. Fill the nebuliser (C) with the volume of the medicinal product as specified in the patient instructions. Attach the mouthpiece of the nebuliser to the inhalation filter using a mouthpiece adapter if required, ensuring that connections are airtight. Make sure the nebuliser is positioned in the same orientation as intended for use; this may require tilting the breathing simulator and filter holder. Set the breathing simulator to generate the specified breathing pattern.

A. inhalation filter and filter holder | B. breathing simulator | C. nebuliser |
Start the breathing simulator then, at the beginning of an inhalation cycle, start the nebuliser. Operate the nebuliser for a defined initial time period. The time chosen, usually 60 ± 1 s, must allow sufficient active substance deposition on the inhalation filter to allow quantitative analysis. If the quantity of active substance deposited on the inhalation filter in 60 s is insufficient for this analysis, the length of the time interval for aerosol collection can be increased. If the filter is soaked with the preparation, this time can be decreased. At the end of this initial period, stop the nebuliser.
Place a fresh filter and filter holder in position and continue until nebulisation ceases. Interrupt nebulisation and exchange filters if necessary, to avoid filter saturation.
Results
Using a suitable method of analysis, determine the mass of active substance collected on the filters and filter holders during each time interval. Determine the active substance delivery rate by dividing the mass of active substance collected on the first inhalation filter by the time interval used for collection. Determine the total mass of active substance delivered by summing the mass of active substance collected on all inhalation filters and filter holders.
AERODYNAMIC ASSESSMENT OF NEBULISED AEROSOLS
Nebulised products need to be size-characterised at flow rates lower than the range that is normally used for powder inhalers and metered-dose inhalers. A flow rate of 15 L/min is recommended in the European standard because this value represents a good approximation to the mid-inhalation flow rate achievable by a tidally breathing healthy adult (500 mL tidal volume).
Although low-angle laser light scattering instruments (laser diffractometers) can provide rapid size-distribution measurements of nebuliser-generated aerosols, these techniques do not detect the active substance; rather they measure the size distribution of the droplets irrespective of their content. This may not be a problem with homogeneous solutions, but can result in significant error if the product to be nebulised is a suspension, or if droplet evaporation is significant as can be the case with certain nebuliser types. Cascade impactors enable the aerosol to be characterised unambiguously in terms of the mass of active substance as a function of aerodynamic diameter. Laser diffraction may be used if validated against a cascade impaction method.
Apparatus E (see below under Apparatus), a cascade impactor, has been calibrated at 15 L/min specifically to meet the recommendation of the European standard, and is therefore used for this test. Determining mass balance in the same way as for powder inhalers and metered-dose inhalers is not straightforward, in that the dose is being captured as a continuous output, and hence is not included. As part of method development, recovery experiments must be performed to validate the method.
It is also recognised that the control of evaporation of droplets produced by nebulisers may be critical to avoid bias in the droplet size assessment process. Evaporation can be minimised by cooling the impactor to a temperature of about 5 °C, typically achieved by cooling the impactor in a refrigerator for about 90 min. Typically, at least after each day of use, the apparatus must be fully cleaned, including the inter-stage passageways, in view of the greater risk of corrosion caused by the condensation/accumulation of saline-containing droplets on inter-stage metalwork associated with cooling the impactor. It is recommended to dry all surfaces of the apparatus after each test, for example with compressed air. Note: the micro-orifice collector (MOC) should not be dried with compressed air.
Apparatus
A detailed description of Apparatus E and the induction port is contained in general chapter 2.9.18, and includes details of critical dimensions and the qualification process for the impactor (stage mensuration).
A back-up filter in addition to the micro-orifice collector (MOC) must be used to ensure quantitative recovery of active substance from the nebulised aerosol at the specified flow rate of 15 L/min. The filter is located below the MOC (internal filter option) or a filter in holder, external to the impactor, is used to capture any fine droplets that pass beyond the last size fractionating stage.
A pre-separator is not used for testing nebuliser-generated aerosols.
Method validation
Impactor stage overloading
During method development and validation, it is important to confirm that the volume of liquid sampled from the nebuliser does not overload the impactor. Visual inspection of the collection surfaces on stages collecting most of the droplets may reveal streaking if overloading has occurred. This phenomenon is usually also associated with an increase in mass of active substance collected on the final stage and back-up filter. Reducing the sampling period (T0) is the most effective way to avoid overloading in any given system, balancing overloading with analytical sensitivity.
Re-entrainment
Droplet bounce and re-entrainment are less likely with nebuliser-produced droplets than with solid particles from inhalers and for that reason coating would not normally be required.
Method
Pre-cool the assembled impactor and induction port in a refrigerator (set at about 5 °C) for not less than 90 min and start the determination within about 5 min of removal of the impactor from the refrigerator. Other methods that maintain the impactor at a constant temperature (for example, use of a cooling cabinet) can also be employed when validated.
Set up the nebuliser with a supply of driving gas (usually air or oxygen), or use a compressor, at the pressure and flow rate specified by the manufacturer of the nebuliser. Take precautions to ensure that the gas supply line does not become detached from the nebuliser when under pressure. Fill the nebuliser with the volume of the medicinal product as specified in the patient instructions.
Remove the impactor from the refrigerator. Attach the induction port to the impactor, and connect the outlet of the impactor/external filter to a vacuum source that is capable of drawing air through the system at 15 L/min as specified in Figure 2.9.44.-2. Turn on the flow through the impactor.

A. nebuliser | B. inducation port | C. impactor (apparatus E) | D. flow control valve | E. vacuum source |
Connect a flow meter, calibrated for the volumetric flow leaving the meter, to the induction port. Adjust the flow control valve located between the impactor and the vacuum source to achieve a steady flow through the system at 15 L/min (± 5 per cent). Remove the flow meter.
Make sure the nebuliser is positioned in the same orientation as intended for use then attach the mouthpiece of the nebuliser to the induction port, using a mouthpiece adapter if required, ensuring that connections are airtight. Switch on the flow/compressor for the nebuliser. Sample for a predetermined time (T0). Once determined, this time (T0) must be defined and used in the analytical method for a particular medicinal product to ensure that mass fraction data can be compared. At the end of the sampling period, switch off the driving gas flow/compressor to the nebuliser, remove the nebuliser from the induction port and switch off the flow from the vacuum source to the impactor.
Dismantle the impactor and, using a suitable method of analysis, determine the mass of active substance collected in the induction port, on each stage and on the back-up filter/external filter as described for Apparatus E in general chapter 2.9.18. Add the mass of active substance collected in the MOC to that deposited on the back-up filter/external filter and treat as a single sample for the purpose of subsequent calculations.
Calculate the mass fraction (Fm,comp) of the active substance deposited on each component of the impactor, commencing with the induction port and proceeding in order through the impactor, using the following expression:

mcomp | = | mass associated with the component under evaluation; |
M | = | total mass collected by the system. |
Present Fm,comp in order of location within the measurement equipment, beginning at the induction port and ending with the back-up filter of the impactor (see Figure 2.9.44.-3). Where appropriate, Fm,comp for adjacent stages of the impactor may be combined in order to report the mass fraction collected on a group of stages as a single value.
Determine the cumulative mass-weighted particle-size distribution of the aerosol size-fractionated by the impactor in accordance with the procedure given in general chapter 2.9.18. Starting at the filter, derive a cumulative mass versus effective cut-off diameter of the respective stages (see Table 2.9.44.-2 for the appropriate cut-off diameters at 15 L/min). Plot the cumulative fraction of active substance versus cut-off diameter in a suitable format, for example logarithmic or log-probability format. Where appropriate, determine by interpolation the fraction either below a given size or between an upper and a lower size limit.

If necessary, and where appropriate, determine values for the mass median aerodynamic diameter (MMAD) and the geometric standard deviation (GSD), as appropriate.

9. Demonstration of Uniformity of Dosage Units Using Large Samples Sizes
The procedure is intended for, but not limited to, the evaluation of medicinal products that are manufactured using process analytical technology (PAT) methodology.
Compliance with general chapter 2.9.40. Uniformity of dosage units can be demonstrated by the following procedure, when large samples (sample size n ≥ 100) are evaluated.
Application of this chapter does not constitute a mandatory requirement. It presents 2 alternative tests (Alternative 1 and Alternative 2). Fulfilling the requirements of either of the 2 alternatives is considered as evidence that the medicinal product tested complies with general chapter 2.9.40. The 2 alternatives are considered equivalent in their demonstration of compliance with general chapter 2.9.40.
ALTERNATIVE 1 (PARAMETRIC)
Select not fewer than 100 units according to a predefined sampling plan.
The consistency of dosage units is evaluated by content uniformity or mass variation as prescribed in Table 2.9.40.-1. Calculate the acceptance value (AV) using the following expression:

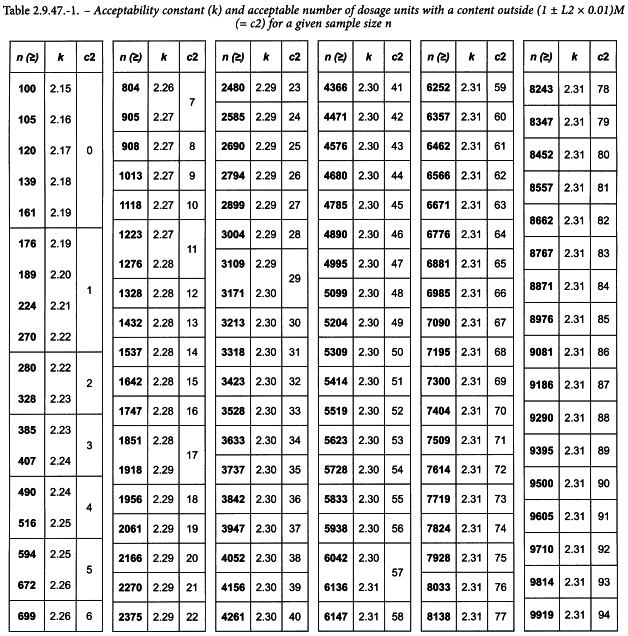
for which the terms are defined in Table 2.9.40.-2, but using the sample size-dependent value for k defined in Table 2.9.47.-1.
Criteria
Apply the following criteria, unless otherwise specified.
The requirements for dosage form uniformity are met if:
1. | the acceptance value (AV) is less than or equal to L1; and |
2. | in the calculation of acceptance value (AV) under content uniformity or under mass variation, the number of individual dosage units outside (1 ± L2 × 0.01)M is less than or equal to c2 as defined for a given sample size n in Table 2.9.47.-1. |
Unless otherwise specified, L1 is 15.0 and L2 is 25.0.
Table 2.9.47.-1. is to be interpreted as follows:
ALTERNATIVE 2 (non-PARAMETRIC)
Select not fewer than 100 units according to a predefined sampling plan.
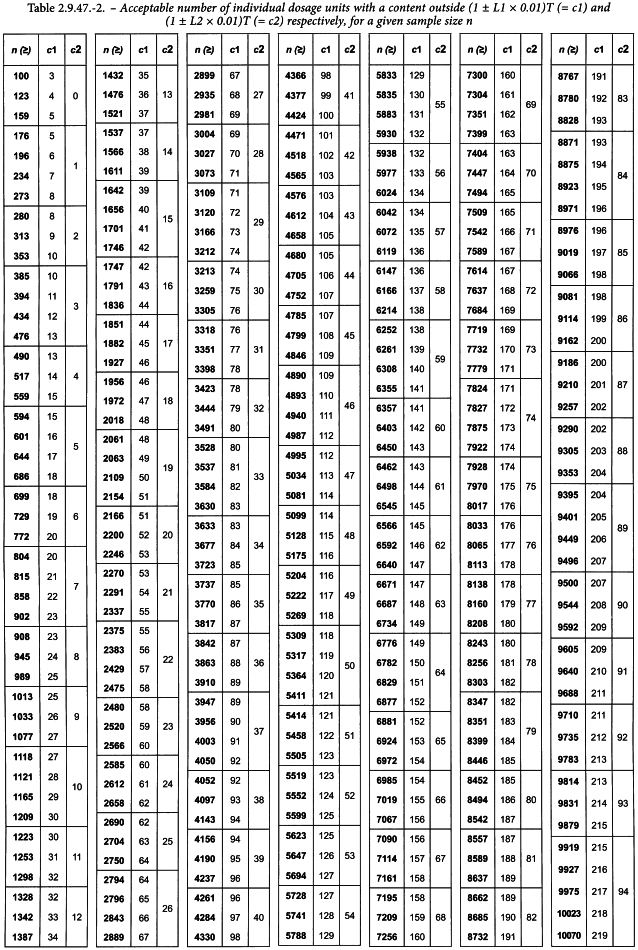
The consistency of dosage units is evaluated by content uniformity or mass variation as prescribed in Table 2.9.40.-1. Assay individually or weigh the units and calculate individual contents as prescribed in general chapter 2.9.40. Count the number of individual dosage units with a content outside (1 ± L1 × 0.01)T and the number of individual dosage units with a content outside (1 ± L2 × 0.01)T. Evaluate if the values are within the limits defined in Table 2.9.47.-2.
Criteria
Apply the following criteria, unless otherwise specified.
The requirements for dosage form uniformity are met if:
1. | the number of individual dosage units outside (1 ± L1 × 0.01)T is less than or equal to c1; and |
2. | the number of individual dosage units outside (1 ± L2 × 0.01)T is less than or equal to c2. |
c1 and c2 for a given sample size n are defined in Table 2.9.47.-2. Unless otherwise specified, L1 is 15.0 and L2 is 25.0.
Table 2.9.47.-2 is to be interpreted as follows:
10. Content of active ingredient on actuation of the valve test
Content of active ingredient delivered by actuation of the valve
Remove the pressurised container from the actuator and remove all labels and markings which may be present on the container with a suitable solvent. Dry the container, replace in its actuator, shake for about 30 seconds and prime the metering valve as follows. Discharge once to waste, wait for not less than 5 seconds and discharge again to waste. Remove the pressurised container from its actuator, clean the valve stem (internally and externally) and the valve ferrule by washing with a suitable solvent. Dry the complete valve assembly using an air line fitted with an appropriate narrow jet to ensure that all solvent is removed from the inside of the valve stem.
Place a stainless steel base plate that has three legs and a central circular indentation with a hole about 1.5 mm in diameter in a small vessel suitable for shaking and add the volume of solvent specified in the monograph. The size of the vessel is such that when the pressurised inhalation is discharged into the specified volume of solvent as described below the discharge takes place not less than 25 mm below the surface of the solvent.
Shake the pressurised container for about 30 seconds and place it inverted in the vessel. Discharge 10 deliveries below the surface of the solvent actuating the valve at intervals of not less than 5 seconds, maintaining the pressurised container in the vertical plane and discharging the pressurised inhalation through the hole in the centre of the base plate. (It may be necessary because of the nature of the formulation to shake the pressurised container between each actuation of the valve; where this is the case shaking should be carried out without removing the pressurised container from its inverted position in the vessel.) Remove the pressurised container, wash it with the specified solvent and dilute the combined solution and washings to the volume specified in the monograph. Determine the amount of active ingredient by the method described under the Assay and calculate the amount delivered from each actuation of the valve. The result lies within the range for the content of active ingredient stated in the monograph.