Appendix XIV C. Test for Bacterial Endotoxins (LAL Test)1
The test for bacterial endotoxins (BET) is used to detect or quantify endotoxins from gram-negative bacteria using amoebocyte lysate from the horseshoe crab (Limulus polyphemus or Tachypleus tridentatus). There are 3 techniques for this test: the gel-clot technique, which is based on gel formation; the turbidimetric technique, based on the development of turbidity after cleavage of an endogenous substrate; and the chromogenic technique, based on the development of colour after cleavage of a synthetic peptide-chromogen complex.
The following 6 methods are described in the present chapter:
Method A. | Gel-clot method: limit test |
Method B. | Gel-clot method: quantitative test |
Method C. | Turbidimetric kinetic method |
Method D. | Chromogenic kinetic method |
Method E. | Chromogenic end-point method |
Method F. | Turbidimetric end-point method |
Proceed by any of the 6 methods for the test. In the event of doubt or dispute, the final decision is made based upon method A unless otherwise indicated in the monograph.
The test is carried out in a manner that avoids endotoxin contamination.
1. apparatus
Depyrogenate all glassware and other heat-stable apparatus in a hot-air oven using a validated process. A commonly used minimum time and temperature is 30 min at 250 °C. If employing plastic apparatus, such as microtitre plates and pipette tips for automatic pipetters, use apparatus shown to be free of detectable endotoxin and which does not interfere in the test.
NOTE: in this chapter, the term ‘tube’ includes all types of receptacles, for example microtitre plate wells.
2. reagents, test solutions
(1) Amoebocyte lysate
Amoebocyte lysate is a lyophilised product obtained from amoebocyte lysate from the horseshoe crab (Limulus polyphemus or Tachypleus tridentatus). This reagent refers only to a product manufactured in accordance with the regulations of the competent authority.
NOTE: amoebocyte lysate reacts with some β-glucans in addition to endotoxins. Amoebocyte lysate preparations which do not react with glucans are available; they are prepared by removing from amoebocyte lysate the G factor, which reacts with glucans, or by inhibiting the G factor reacting system of amoebocyte lysate. These preparations may be used for endotoxin testing in the presence of glucans.
(2) Lysate solution
Dissolve amoebocyte lysate in water for BET or in a buffer, as recommended by the lysate manufacturer, by gentle stirring. Store the reconstituted lysate, refrigerated or frozen, as indicated by the manufacturer.
(3) Water for BET (water for bacterial endotoxins test)
Water for injections R or water produced by other procedures that shows no reaction with the lysate employed at the detection limit of the reagent.
3. Preparation of the standard endotoxin stock solution
The standard endotoxin stock solution is prepared from an endotoxin reference standard that has been calibrated against the International Standard, for example endotoxin standard BRP.
Endotoxin is expressed in International Units (IU). The equivalence in IU of the International Standard is stated by the World Health Organization.
NOTE: one International Unit (IU) of endotoxin is equal to one Endotoxin Unit (E.U.).
Follow the specifications in the package leaflet and on the label for preparation and storage of the standard endotoxin stock solution.
4. Preparation of the standard endotoxin solutions
After vigorously mixing the standard endotoxin stock solution, prepare appropriate serial dilutions of this solution using water for BET.
Use the solutions as soon as possible to avoid loss of activity by adsorption.
5. Preparation of the test solutions
Prepare the test solutions by dissolving or diluting active substances or medicinal products using water for BET. Some substances or preparations may be more appropriately dissolved or diluted in other aqueous solutions. If necessary, adjust the pH of the test solution (or dilution thereof) so that the pH of the mixture of the lysate and test solution falls within the pH range specified by the lysate manufacturer, usually 6.0 to 8.0. The pH may be adjusted by the use of acid, base or a suitable buffer, as recommended by the lysate manufacturer. Acids and bases may be prepared from concentrates or solids with water for BET in containers free of detectable endotoxin. Buffers must be validated to be free of detectable endotoxin and interfering factors.
6. Determination of the Maximum Valid Dilution
The Maximum Valid Dilution (MVD) is the maximum allowable dilution of a sample at which the endotoxin limit can be determined. Determine the MVD using the following formulae:
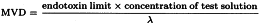
Endotoxin limit The endotoxin limit for active substances administered parenterally, defined on the basis of dose, is equal to:
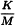
K | = | threshold pyrogenic dose of endotoxin per kilogram of body mass, |
M | = | maximum recommended bolus dose of product per kilogram of body mass. |
When the product is to be injected at frequent intervals or infused continuously, M is the maximum total dose administered in a single hour period.
The endotoxin limit for active substances administered parenterally is specified in units such as IU/mL, IU/mg, IU/Unit of biological activity, etc., in monographs.
λ | = | the labelled lysate sensitivity in the gel-clot technique (IU/mL) or the lowest concentration used in the standard curve of the turbidimetric or chromogenic techniques. |
7. GEL-CLOT TECHNIQUE (METHODS A AND B)
The gel-clot technique allows detection or quantification of endotoxins and is based on clotting of the lysate in the presence of endotoxins. The minimum concentration of endotoxins required to cause the lysate to clot under standard conditions is the labelled lysate sensitivity. To ensure both the precision and validity of the test, confirm the labelled lysate sensitivity and perform the test for interfering factors as described under 1. Preparatory testing.
1. Preparatory testing
(i) Confirmation of the labelled lysate sensitivity
Confirm in 4 replicates the labelled sensitivity λ, expressed in IU/mL, of the lysate solution prior to use in the test. Confirmation of the lysate sensitivity is carried out when a new lot of lysate is used or when there is any change in the test conditions which may affect the outcome of the test.
Prepare standard solutions of at least 4 concentrations equivalent to 2λ, λ, 0.5λ and 0.25λ by diluting the standard endotoxin stock solution with water for BET.
Mix a volume of the lysate solution with an equal volume of 1 of the standard solutions (such as 0.1 mL aliquots) in each tube. When single test vials or ampoules containing lyophilised lysate are employed, add solutions of standards directly to the vial or ampoule. Incubate the reaction mixture for a constant period according to the recommendations of the lysate manufacturer (usually at 37 ± 1 °C for 60 ± 2 min), avoiding vibration. Test the integrity of the gel: for tubes, take each tube in turn directly from the incubator and invert it through approximately 180° in one smooth motion. If a firm gel has formed that remains in place upon inversion, record the result as positive. A result is negative if an intact gel is not formed.
The test is considered valid when the lowest concentration of the standard solutions shows a negative result in all replicate tests.
The end-point is the lowest concentration in the series of decreasing concentrations of standard endotoxin that clots the lysate. Determine the geometric mean end-point concentration by calculting the mean of the logarithms of the end-point concentrations of the 4 dilution series, take the antilogarithm of this value, as indicated by the following expression:
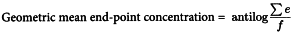
Σe | = | sum of the log10 end-point concentrations of the dilution series used, |
f | = | number of replicates. |
The geometric mean end-point concentration is the measured sensitivity of the lysate solution (IU/mL). If this is not less than 0.5λ and not more than 2λ, the labelled sensitivity is confirmed and is used in the tests performed with this lysate.
(ii) Test for interfering factors
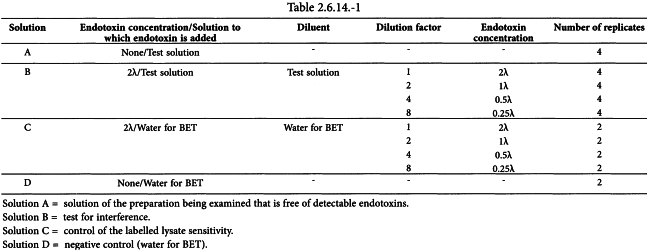
Prepare solutions A, B, C and D as shown in Table 2.6.14.-1, and use the test solutions at a dilution less than the MVD, not containing any detectable endotoxins, operating as described under 1. Preparatory testing, (i) Confirmation of the labelled lysate sensitivity.
The geometric mean end-point concentrations of solutions B and C are determined using the expression described in 1. Preparatory testing, (i) Confirmation of the labelled lysate sensitivity.
The test for interfering factors must be repeated when any changes are made to the experimental conditions that are likely to influence the result of the test.
The test is considered valid when all replicates of solutions A and D show no reaction and the result of solution C confirms the labelled lysate sensitivity.
If the sensitivity of the lysate determined with solution B is not less than 0.5λ and not greater than 2λ, the test solution does not contain interfering factors under the experimental conditions used. Otherwise, the test solution interferes with the test.
If the preparation being examined interferes with the test at a dilution less than the MVD, repeat the test for interfering factors using a greater dilution, not exceeding the MVD. The use of a more sensitive lysate permits a greater dilution of the preparation being examined and this may contribute to the elimination of interference.
Interference may be overcome by suitable validated treatment, such as filtration, neutralisation, dialysis or heat treatment. To establish that the treatment chosen effectively eliminates interference without loss of endotoxins, repeat the test for interfering factors using the preparation being examined to which the standard endotoxin has been added and which has then been submitted to the chosen treatment.
2. Limit test (method a)
(i) Procedure
Prepare solutions A, B, C and D as shown in Table 2.6.14.-2, and perform the test on these solutions following the procedure described under 1. Preparatory testing, (i) Confirmation of the labelled lysate sensitivity.

Prepare solution A and solution B (positive product control) using a dilution not greater than the MVD and treatments as described in 1. Preparatory testing, (ii) Test for interfering factors. Solutions B and C (positive controls) contain the standard endotoxin at a concentration corresponding to twice the labelled lysate sensitivity. Solution D (negative control) consists of water for BET.
(ii) Interpretation
The test is considered valid when both replicates of solution B and C are positive and those of solution D are negative.
When a negative result is found for both replicates of solution A, the preparation being examined complies with the test.
When a positive result is found for both replicates of solution A, the preparation being examined does not comply with the test.
When a positive result is found for one replicate of solution A and a negative result is found for the other, repeat the test. In the repeat test, the preparation being examined complies with the test if a negative result is found for both replicates of solution A. The preparation does not comply with the test if a positive result is found for one or both replicates of solution A.
However, if the preparation does not comply with the test at a dilution less than the MVD, the test may be repeated using a greater dilution, not exceeding the MVD.
3. quantitative test (method b)
(i) Procedure
The test quantifies bacterial endotoxins in the test solution by titration to an end-point. Prepare solutions A, B, C and D as shown in Table 2.6.14.-3, and test these solutions according to the procedure described under 1. Preparatory testing, (i) Confirmation of the labelled lysate sensitivity.
(ii) Calculation and interpretation
The test is considered valid when the following 3 conditions are met:
(a) both replicates of solution D (negative control) are negative,
(b) both replicates of solution B (positive product control) are positive,
(c) the geometric mean end-point concentration of solution C is in the range of 0.5λ to 2λ.
To determine the endotoxin concentration of solution A, calculate the end-point concentration for each replicate, by multiplying each end-point dilution factor by λ.
The endotoxin concentration in the test solution is the end-point concentration of the replicates. If the test is conducted with a diluted test solution, calculate the concentration of endotoxin in the original solution by multiplying the result by the dilution factor.
If none of the dilutions of the test solution is positive in a valid test, report the endotoxin concentration as less than λ (or, if a diluted sample was tested, report as less than the lowest dilution factor of the sample × λ). If all dilutions are positive, the endotoxin concentration is reported as equal to or greater than the largest dilution factor multiplied by λ (e.g. in Table 2.6.14.-3, the initial dilution factor × 8 × λ).
The preparation being examined meets the requirements of the test if the endotoxin concentration in both replicates is less than that specified in the monograph.

8. PHOTOMETRIC quantitative TECHNIQUES (METHODS C, D, E AND F)
1. Turbidimetric technique (methods c and f)
This technique is a photometric test to measure the increase in turbidity. Based on the test principle employed, this technique may be classified as being either the end-point-turbidimetric test or the kinetic-turbidimetric test.
The end-point-turbidimetric test (Method F) is based on the quantitative relationship between the endotoxin concentration and the turbidity (absorbance or transmission) of the reaction mixture at the end of an incubation period.
The kinetic-turbidimetric test (Method C) is a method to measure either the time (onset time) needed for the reaction mixture to reach a predetermined absorbance or transmission, or the rate of turbidity development.
The test is carried out at the incubation temperature recommended by the lysate manufacturer (usually 37 ± 1 °C).
2. Chromogenic technique (methods d and e)
This technique is used to measure the chromophore released from a suitable chromogenic peptide by the reaction of endotoxins with the lysate. Depending on the test principle employed, this technique may be classified as being either the end-point-chromogenic test or the kinetic-chromogenic test.
The end-point-chromogenic test (Method E) is based on the quantitative relationship between the endotoxin concentration and the quantity of chromophore released at the end of an incubation period.
The kinetic-chromogenic test (Method D) measures either the time (onset time) needed for the reaction mixture to reach a predetermined absorbance, or the rate of colour development.
The test is carried out at the incubation temperature recommended by the lysate manufacturer (usually 37 ± 1 °C).
3. Preparatory testing
To assure the precision or validity of the turbidimetric and chromogenic techniques, preparatory tests are conducted to show that the criteria for the standard curve are satisfied and that the test solution does not interfere with the test.
Validation of the test method is required when any changes are made to the experimental conditions that are likely to influence the result of the test.
(i) Assurance of criteria for the standard curve
The test must be carried out for each lot of lysate reagent.
Using the standard endotoxin solution, prepare at least 3 endotoxin concentrations within the range indicated by the lysate manufacturer to generate the standard curve. Perform the test using at least 3 replicates of each standard endotoxin solution as recommended by the lysate manufacturer (volume ratios, incubation time, temperature, pH, etc.).
If the desired range is greater than 2 log10 in the kinetic methods, additional standards must be included to bracket each log10 increase in the range of the standard curve.
The absolute value of the correlation coefficient, | r |, must be greater than or equal to 0.980, for the range of endotoxin concentrations set up.
(ii) Test for interfering factors
Select an endotoxin concentration at or near the middle of the endotoxin standard curve.
Prepare solutions A, B, C and D as shown in Table 2.6.14.-4. Perform the test on at least 2 replicates of these solutions as recommended by the lysate manufacturer (volume of test solution and lysate solution, volume ratio of test solution to lysate solution, incubation time, etc.).
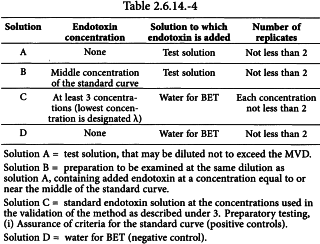
The test is considered valid when the following conditions are met:
Calculate the mean recovery of the added endotoxin by subtracting the mean endotoxin concentration in the solution (if any) (solution A, Table 2.6.14.-4) from that in the solution containing the added endotoxin (solution B, Table 2.6.14.-4).
The test solution is considered free of interfering factors if under the conditions of the test, the measured concentration of the endotoxin added to the test solution is within 50-200 per cent of the known added endotoxin concentration, after subtraction of any endotoxin detected in the solution without added endotoxin.
When the endotoxin recovery is out of the specified range, the test solution is considered to contain interfering factors. Repeat the test using a greater dilution, not exceeding the MVD. Furthermore, interference of the test solution or diluted test solution not to exceed the MVD may be eliminated by suitable validated treatment, such as filtration, neutralisation, dialysis or heat treatment. To establish that the treatment chosen effectively eliminates interference without loss of endotoxins, repeat the test for interfering factors using the preparation being examined to which the standard endotoxin has been added and which has then been submitted to the chosen treatment.
4. Test
(i) Procedure
Follow the procedure described in 3. Preparatory testing, (ii) Test for interfering factors.
(ii) Calculation
Calculate the endotoxin concentration of each replicate of solution A using the standard curve generated by the positive control solution C.
The test is considered valid when the following 3 requirements are met:
(1) the results obtained with solution C comply with the requirements for validation defined under 3. Preparatory testing, (i) Assurance of criteria for the standard curve,
(2) the endotoxin recovery, calculated from the endotoxin concentration found in solution B after subtracting the endotoxin concentration found in solution A, is within the range of 50-200 per cent,
(3) the result obtained with solution D (negative control) does not exceed the limit of the blank value required in the description of the lysate employed, or it is less than the endotoxin detection limit of the lysate reagent employed.
(iii) Interpretation
The preparation being examined complies with the test if the mean endotoxin concentration of the replicates of solution A, after correction for dilution and concentration, is less than the endotoxin limit for the product.
Guidelines on the test for bacterial endotoxins are given in general chapter 5.1.10.