Appendix XIV O. Host-cell Protein Assays
INTRODUCTION
Host-cell proteins (HCPs) are process-related impurities derived from the host organism used for the production of a medicinal product by recombinant DNA technology. In order to mitigate their potential adverse effects (e.g. immunogenicity), HCP content is expected to be reduced to the lowest possible level.
HCP clearance during the purification process must be assessed and the HCP content determined using an HCP assay that has been evaluated and validated for a given product.
The HCP acceptance limit, typically expressed in nanograms of HCP per milligram of active substance (ppm), must be justified with regard to the HCP clearance capacity of the purification process and with regard to the potential impact of residual HCP on patients, taking into account the worst-case quantity of HCP that could be administered with the product.
HCPs are generally measured using an immuno-based assay containing, as reagents, the HCP antigen preparation (hereinafter ‘the HCP antigens’) or HCP reference standard and the corresponding polyclonal antibodies (antisera). Antisera must cover a broad spectrum of HCPs representative of the product concerned.
Sandwich-type enzyme-linked immunosorbent assays (ELISA) are the most commonly employed assays to assess quantitatively the level of HCPs. It should be noted that HCP content measured by ELISA does not represent absolute HCP mass content. The sensitivity is the result of the observed cumulative responses of many individual HCPs in comparison to the response of an HCP reference standard. The use of orthogonal analytical methods (e.g. electrophoresis, HPLC, Western blot, mass spectrometry) to characterise the various HCPs in the product is recommended to support the development and selection of the assay.
ASSAY SELECTION
Several types of assay are available, with selection taking into account several factors, including the stage of development of the product, the nature of the host cell and the protein immunogenicity, the expression mode, the manufacturing process, and prior knowledge. When selecting and developing the assay, its life cycle (e.g. reagent supply, consistency, assay validation, process change) must also be considered.
Types of assay
Process-specific assays
Process-specific HCP assays (also called product-specific HCP assays) are developed and validated taking into account the specificity of the production process, and using the same host organism expressing the recombinant product.
The HCP antigens are derived from a mock run of the active substance manufacturing process (or a process representative of it) up to a step capable of generating a broad spectrum of HCPs in sufficient quantities.
The antisera raised must cover a broad range of HCPs, in order to detect as many different HCPs as possible and also to accommodate process variations.
Platform assays
Platform assays are developed by individual manufacturers and customised for the processes and host organism used by the manufacturer for production. The same sets of reference standards and reagents may be used to monitor HCPs in several products manufactured in the same host organism, provided that upstream processes (and downstream, if relevant) are sufficiently similar for these products. The suitability of the antiserum should be evaluated as described above for process-specific assays.
Generic assays
Commercially available HCP test kits are commonly referred to as generic HCP assays. They are intended to work broadly across similar expression hosts. Detailed information on the preparation of the reagents may not be disclosed by the vendor. For instance, the HCP antigens may be derived from a combination of strains of an expression host species, and the process(es) used may not mimic the process applied for the product of interest. The suitability of the antiserum should be evaluated as described above for process-specific assays.
Criteria for assay selection
In view of the potential safety issues associated with residual HCPs in the active substance, a risk assessment is performed to support the choice between a generic, a platform or a process-specific strategy, taking into account the stage of development of the product.
For early development a generic assay or a platform assay may be used. For later development phases, process-specific assays must be considered, as they are generally regarded as superior, especially when compared to generic assays. This is because process-specific assays are more likely to show immunoreactivity against representative HCPs.
Platform or generic assays may be used, provided that the assay is appropriately characterised and validated against process-specific HCPs.
PRODUCTION AND TESTING OF THE HCP ANTIGEN preparation
The HCP reagents (HCP antigens and anti-HCP antibodies) are produced in such a way as to facilitate replication of the production when a replenishment for the HCP assay is needed.
The HCP antigens are used to generate the polyclonal antibody reagent for the HCP immunoassay by immunising one or more suitable animal species. In addition, they serve as the HCP reference standard in the HCP immunoassay.
As far as possible, the HCP antigens must cover the relevant HCP population expected to be derived from the manufacturing process of the protein of interest.
The HCP population must also be broad enough to cover worst-case purification scenarios and to provide robustness against potential manufacturing process changes during the life cycle of the product.
Process-Specific Assays
Null cell line
Development of a process-specific assay involves the selection of a null cell line that does not contain the expression gene for the product of interest and is derived from the same cell line that has been used to establish the production cell line. This null cell line may be non-transfected or mock-transfected. A mock-transfected cell line is created by transfecting the parental cell line with a blank plasmid, i.e. the plasmid used to create the production cell line, but missing the gene coding for the protein of interest.
Mock production process
Upstream
The antigens produced for process-specific assays are obtained by a mock production process that mimics the intended manufacturing process, using the null cell line and, as far as possible, the same operating conditions.
As for any mock production, the process used represents an approximation of the intended manufacturing process and leads to differences (e.g. different scale, operating parameters, product interaction). However, the impact of those differences needs to be considered carefully because they may affect the composition of the HCP population.
For example, a mock fermentation of an inclusion body manufacturing process may not deliver the desired inclusion bodies if the product is not present. Therefore, depending on the null cell line used (e.g. mock-transfected or not), the antigens may need to be isolated differently compared to the intended manufacturing process.
In some situations, operating parameters for the mock production may be adjusted to cover worst-case scenarios (e.g. to deliver antigens covering a broad spectrum of different HCP species). For example, the antigen-containing cell culture supernatant may be harvested beyond the minimum level of cell viability in order to include more cytosolic proteins, which are released by additional cell lysis.
Downstream
The HCP antigens derived from the upstream process are usually only minimally processed (filtration, concentration), in order to obtain a representative spectrum of HCPs. Further purification is generally not recommended as there will be a risk of losing HCP species.
However, in cases where the antigens are not representative (e.g. resulting in low coverage), mixing of mock materials from different processing steps can be considered. Enrichment may also be achieved by pooling materials from mock fermentation or purification runs using different operating conditions, or from selective purification steps (e.g. to reduce large amounts of the few immunodominant HCPs).
Cross-contamination with the protein of interest
The HCP antigens must be produced in a manner that avoids contamination with even minute traces of the product in order to avoid cross-reactivity with the polyclonal antibodies.
To achieve this goal, dedicated or single-use equipment is used as much as possible. Where multi-purpose equipment is used, it must be cleaned appropriately. In addition, the risk of contamination when filling or handling the antigens in the laboratory environment must also be considered.
Characterisation and testing
Before using the HCP antigens for immunisation, the protein content is assessed (total protein assay) and the absence of the protein of interest verified.
Comparison of the HCP population with the mock and the intended production process is performed, typically by SDS-PAGE and/or two-dimensional (2D) electrophoresis with a high sensitivity stain. The aim of this comparison is to show that the HCP antigens resulting from the mock production process contain most of the representative HCP species of the intended manufacturing process. Where necessary, complementary information may be gathered by orthogonal methods, e.g. mass spectrometry.
Platform Assays
Null cell line
Development of a platform assay involves a null cell line that does not contain the expression gene for the product of interest, and uses the same host species. This null cell line may be non-transfected or mock-transfected, and may be used for the production of HCP antigens for products from a given company′s manufacturing platform.
Mock production process
Upstream
The HCP antigens produced for platform assays are obtained by a mock production process that mimics the platform upstream process that is used for several products, and typically uses the same media components. As for any mock production, the process used represents an approximation of the intended manufacturing process, which may impact the composition of the HCP population (see process-specific assays).
Downstream
As for other assays, the HCP antigens derived from the upstream process are, in general, only minimally processed (e.g. no or limited number of purification steps) to obtain a broad spectrum of HCPs, although mixing and pooling strategies may also be used to widen the spectrum of HCP species.
Characterisation and testing
As for process-specific assays, both the protein content and the absence of the protein of interest are tested. Comparison of the HCP population with the mock and the intended production process is performed.
Generic Assays
Generic assays are commercially available and are developed by the vendor.
Detailed information on the preparation of the reagents may not be disclosed by the vendor. For instance, the null cell line may be derived from a combination of strains of an expression host species, and the process(es) used may not mimic the process applied for the product of interest.
Nevertheless, the generic assay must be selected with consideration given to the intended manufacturing process (e.g. appropriate host cell line), and be appropriately validated for the product of interest and phase of development. As a consequence, if generic assays are used in later stages of development or during commercial manufacturing, it is recommended to validate the assay and control lot-to-lot reagent consistency using either appropriate upstream fractions from the production process or a mock preparation generated using a null cell line.
PRODUCTION AND CHARACTERISATION OF THE ANTI-HCP ANTIBODY REAGENT
Process-Specific and Platform Assays
Immunisation
One of the challenges of the immunisation step is to generate polyclonal antibodies that are highly specific and sensitive for each of the antigenic proteins in the complex mixture of HCPs used as an immunogen. An animal’s immune response must be stimulated against both the stronger and the weaker antigens.
An animal host that yields a sufficient quantity and diversity of HCP-specific immunoglobulin G (IgG) is selected.
Where both the polyclonal capture and the polyclonal detection antibodies are from the same source, it can be assumed that they recognise different epitopes on the same HCP in the assay. Alternatively, polyclonal anti-HCP antibodies from different animal species may be used. Using several animals for a given species may reduce the impact of individual variations in immune competence and provide additional response diversity, resulting in maximised antibody coverage against the HCP antigens.
An immune response to a limited number of HCP antigens may be obtained rapidly, particularly when adjuvants are used to boost the immune response. However, in complex mixtures, differential enhancement of the immune response towards weaker antigens or those at lower concentrations may be necessary.
It usually takes several immunisations to reach a maximum immunological response and, depending on the frequency of immunisation, the process can take 3-6 months to complete.
The immune response against the HCPs for a given immunisation scheme has to be monitored by determining the antibody titre using, for example, an ELISA, and by comparing the results of 1D or 2D electrophoresis after protein staining and a Western blot, where the polyclonal anti-HCP antibodies are used as primary antibody. In practice, some minor proteins that elicit a strong immune response may not be visible in the protein-stained gel, and some poorly antigenic proteins that are detectable by protein staining may not elicit a detectable immune response. To achieve sample-dilution linearity in complex multi-analyte immunoassays, it is essential that the immune reagent simultaneously and specifically recognises as many individual analytes as possible in an assay sample and that it is present in stoichiometric excess. For this purpose, a series of sample dilutions from different process steps may be tested by ELISA using purified anti-HCP antibodies from bleedings that have shown suitable coverage by Western blot.
Finally, based on the results of the tests described above, antisera from different animals are pooled, retested and purified.
Purification and preparation
The HCP antibodies must be purified before an assay can be developed.
Typically, this is achieved by protein A- or protein G-chromatography and/or HCP antigen affinity chromatography. In the case of HCP antigen affinity chromatography, the antigens used for immunisation are immobilised on column chromatography media and the specific antibodies are captured by applying the antisera onto the column.
Additional purification to remove potential aggregates might be required by gel permeation chromatography.
For the ELISA, a part of the purified anti-HCP antibodies is conjugated to a detection label (e.g. biotin or horseradish peroxidase).
The purified anti-HCP antibodies and the sera must be stored at a temperature that ensures their stability.
Characterisation and testing
The suitability of the derived HCP assay reagent is assessed by demonstrating the coverage of the HCPs representative of the manufacturing process by the anti-HCP antibodies.
For this purpose, 2D electrophoresis of the HCP antigens is performed. The protein pattern of the immunostain is compared with the protein pattern of the total stain. The anti-HCP antibodies must recognise a broad range of HCPs over the full range of charge and molecular size. Other methods using native conditions may be considered.
Generic Assays
Immunisation, purification and preparation of the anti-HCP antibody reagent are carried out by the vendor and details may not be available.
Characterisation and testing of anti-HCP antibodies are performed as for other assay types. Typically, there is limited control over lot-to-lot reagent consistency. Appropriate comparative lot testing is therefore required.
VALIDATION OF THE HCP ASSAY
The HCP ELISA is developed to detect and quantify a heterogeneous mixture of antigens at varying concentrations, and with a reagent containing antibodies that are not represented at a one-to-one ratio. The section below is intended to target the specifics in development and validation of this type of ELISA.
HCP assays such as ELISA are validated with regard to accuracy, specificity, precision, quantitation and detection limits, linearity, range and robustness.
During the life cycle of the product, a full or partial revalidation of the assay may be required, for example when implementing a manufacturing process change that may impact the suitability of the HCP reagent.
Accuracy
Accuracy is demonstrated by spike/recovery analysis of the HCP reference standard in a relevant background matrix (e.g. the active substance or a sample from a relevant purification step).
Specificity
Specificity is demonstrated by the absence of interference from the matrix background (including the active substance). For instance, data from the accuracy study can be used to assess specificity.
Precision
As for any other quantitative assay, repeatability, intermediate precision and reproducibility are appropriately demonstrated.
Quantitation and detection limits
Sensitivity is usually in the ppm range and is normally described through the quantitation limit (QL). QL is typically determined by HCP spike recovery studies in the active substance or an appropriate sample matrix, and is calculated from the minimal spike providing a response with predefined accuracy and precision from replicate analyses.
Detection limit (DL) is often not determined (optional validation parameter).
Linearity
The linearity of the HCP assay is demonstrated using dilution series of the HCP standard and spike/recovery experiments (accuracy study).
Additionally, due to the nature of HCP assays, the multiple HCP analytes and polyclonal anti-HCP antibodies, sample-dilution non-linearity may be observed, i.e. back-calculated results increase with increasing dilutions of samples, which in most cases is related to the excess of one or more individual HCPs in the sample when compared to the available antibodies in the HCP immunoassay. As a consequence, dilution linearity must be properly assessed for the relevant process steps by comparison of target versus measured HCP concentrations at varying sample dilutions. Dilution linearity is demonstrated if the acceptance criteria for assay variation are met for different sample dilutions. Studies demonstrating dilution linearity can be carried out either during method development or at the latest during method validation.
If a sample shows dilution non-linearity, multiple sample dilutions are prepared beyond the range where non-linear behaviour is observed.
The final HCP value is typically reported as the average HCP concentration obtained for a minimum of 2 dilutions within the linear dilution range. If justified, 1 dilution may be sufficient.
Range
The range of the assay is typically defined by the HCP concentrations for which a suitable level of precision, accuracy and linearity has been demonstrated.
Robustness
The evaluation of robustness is considered during the development phase.
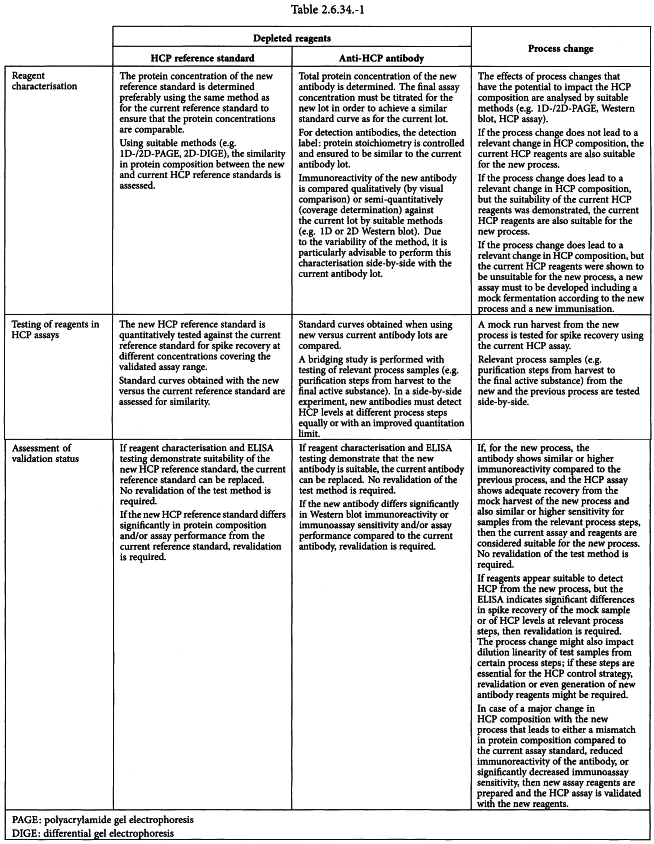
CHANGE OF HCP ASSAY AND/OR REAGENT
The quantities of antigens and antibodies must be large enough to supply the HCP assay for several years. Therefore, the supply, quality and consistency of reagents must be appropriately managed throughout the life cycle of the assay.
For generic HCP assays, in order to ensure the consistency and quality of the reagents, recharacterisation or revalidation of the assay may be required for each new batch of reagent, as their quality may change from one batch to another.
For process-specific and for platform assays, there are generally 2 situations where new HCP assay reagents may be required:
Newly prepared reagents must be thoroughly characterised (e.g. by 2D-SDS-PAGE/Western blot for coverage, 2D-SDS-PAGE/differential gel electrophoresis (DIGE)/identification by MS). Afterwards, the validation status of the assay using the new reagents must be assessed. It is recommended to perform these experiments side-by-side with the currently used reagents.
Table 2.6.34.-1 outlines a recommendation for reagent characterisation and assessment of immunoassay validation, as a consequence of a depletion of assay reagents or a process change.