Appendix XVII V. Characterisation of Crystalline Solids by Microcalorimetry and Solution Calorimetry
INTRODUCTION - THE CONCEPT OF CRYSTALLINITY
The perfectly ordered crystal lattice with every molecule in its expected lattice position is an ideal that is seldom, if ever, achieved. The other extreme is the amorphous state, in which a solid contains the maximum possible density of imperfections (defects of various dimensionalities), such that all long-range order is lost while only the short-range order, imposed by its nearest neighbours, remains. Real crystals lie somewhere between these 2 extremes. A crystal’s position on a scale bounded by these 2 extremes is termed crystallinity.
All real crystals, even in the pure state, possess some lattice imperfections or defects, which increase both the energy (enthalpy under conditions of constant atmospheric pressure) and the disorder (expressed as the entropy) of the crystal lattice. A crystal with a relatively low density of imperfections is said to be highly crystalline and to possess a high crystallinity. By contrast, a particle with a relatively high density of imperfections is said to be partially amorphous and to possess a low crystallinity. In ideal terms, a totally amorphous particle corresponds to zero crystallinity. Amorphous particles may contain somewhat ordered domains that can act as nuclei for crystallisation; such so-called amorphous particles are said to possess a low-level but finite crystallinity.
The ability to detect and to quantify the amount of amorphous material within a highly crystalline substance is of great importance during the development and subsequent manufacture of a pharmaceutical preparation.
In reality, a powder probably contains particles with different degrees of crystallinity, just as it may contain particles with varying sizes and shapes. The lower the crystallinity of a solid, the greater its enthalpy and entropy. The increase in enthalpy is never totally compensated for by the increase in entropy; therefore, the Gibbs free energy, which reflects the balance between them, actually increases. Hence, the lower the crystallinity of a material (powder), and consequently the greater its amorphous character, the greater its apparent intrinsic solubility and dissolution rate, but the lower its thermodynamic stability. Because of the great relevance of these properties, crystallinity is also an important property and requires measurement by a suitable method.
In the following chapter, the crystallinity or the content of amorphous parts of a powder are measured by calorimetric methods such as microcalorimetry or solution calorimetry, although other methods could be used (e.g. see general chapter 2.9.33. Characterisation of crystalline and partially crystalline solids by X-ray powder diffraction (XRPD)).
Many substances are capable of crystallising in more than one type of crystal lattice, which is known as polymorphism. If water or a solvent is incorporated in the crystal lattice the crystals are termed hydrates or solvates. Because of the different crystal packing, and/or molecular conformation and lattice energy, they usually exhibit different physical properties. For simplicity, calorimetry measurements for degree of crystallinity determination discussed here assume only one solid crystalline form present in the material of interest. The theory and experimental technique can be easily expanded to polymorphic systems with proper consideration of the enthalpy differences among the polymorphs.
method 1 - MICROCALORIMETRY (DETERMINation of AMORPHOUS CONTENT)
Most chemical, physical and biological processes are associated with the exchange of heat. Microcalorimetry is a highly sensitive technique to monitor and quantify both exothermic (heat producing) and endothermic (heat absorbing) changes associated with those processes. The technique allows the determination of the rate and extent of chemical reactions, changes of phase or changes of structure.
Thermal events producing only a fraction of a microwatt can be observed using microcalorimetry. This means that temperature differences less than 10-6 K must be detectable. Microcalorimetry typically uses the heat flow (heat leakage) principle, where the heat produced (or absorbed) in a thermally defined vessel flows away (or into) in an effort to re-establish thermal equilibrium with its surroundings. Exceptional thermal stability with its surrounding has to be achieved either by a heat sink or an electronically regulated surrounding.
Heat energy from an active sample in the reaction vessel is channelled typically through Peltier elements; they act as thermoelectric generators using the Seebeck effect. The heat energy is converted into a voltage signal proportional to the heat flow.
Results are typically presented as a measure of the thermal energy produced per unit of time (Watt) as a function of time.
apparatus
Microcalorimeters are typically designed as twin systems with a measuring vessel and a reference vessel. Vessels are typically made of glass or stainless steel. For certain applications specially designed vessels which allow the addition of a gas, a liquid or a solid material may be used.
CALIBRATION
The microcalorimeter is calibrated for heat flow (energy per time unit) using either calibrated external or internal electrical heat sources or a suitable standard reaction.
SENSITIVITY
The sensitivity of the microcalorimetric method can be assessed based on an appropriate standard sample analysed according to the corresponding method in conjunction with the determination of the instrument baseline noise.
PROCEDURE
Weigh in a suitable vessel an appropriate quantity of the substance to be examined. Close the vessel carefully to avoid any evaporation of solvents and place the vessel in the sample holder. If appropriate, allow the vessel to equilibrate at the temperature of the measurement before placing it in the measuring position.
Begin the analysis and record the heat flow, with the time on the abscissa and the heat flow on the ordinate (specify the direction of exothermic or endothermic heat flow).
Detection and quantification of amorphous content in powders
The amorphous state is metastable with respect to the crystalline state; recrystallisation may therefore occur. The measurement of the heat of recrystallisation enables the amorphous content to be determined by the area of the recrystallisation peak. By relating the output from the microcalorimeter for a sample to that obtained from an amorphous standard, it is possible to quantify the amorphous content of the sample. The range of amorphous content covered by this method depends on the individual substance to be tested; in favourable cases limits of detection below 1 per cent can be reached.
Recrystallisation can be initiated by subjecting the sample to higher relative humidity or an atmosphere containing organic vapour. The sample is typically placed in an ampoule which also contains a small test-tube containing an aqueous saturated salt solution, an organic solvent, or a solvent mixture.
The heat of recrystallisation is typically measured using a fixed sample mass placed in a glass or steel vessel. The test-tube containing a saturated salt solution or an organic solvent is chosen to be large enough to allow a full saturation of the atmosphere above the sample. The mass of the sample and the nature of the vapour atmosphere above the sample are chosen so that recrystallisation occurs in such a way that a distinct peak is observed, clearly separated from initial thermal events caused by introduction of the sample.
The conditions under which the transition of the amorphous phase to a thermodynamically more stable crystalline state occurs will have a significant impact on the time of recrystallisation. In particular, physical mixtures of purely amorphous and crystalline material will behave differently from a partially crystalline material. These effects should be considered when developing a method.
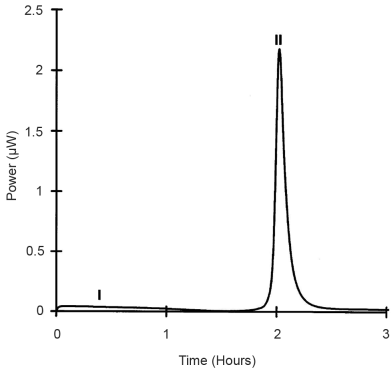
A typical response for the recrystallisation of a mainly amorphous material is shown in Figure 2.2.61.-1. The first part of the curve represents several concurrent processes taking place simultaneously, such as the absorption of water vapour into the amorphous parts of the powder and the generation of water vapour from the test-tube. After this initial response there is a large exothermic response caused by the recrystallisation of the amorphous material. Also included, but not seen, are the expulsion of excess water from the recrystallised parts and its condensation. Thus, the area under this exothermic recrystallisation response is proportional to the heat of recrystallisation.
method 2 - SOLUTION CALORIMETRY (DETERMINation of CRYSTALLINITY)
Solution calorimetry provides a means of determining enthalpy of solution (i.e. heat of solution under constant atmospheric pressure) of a substance. Enthalpy of solution is defined as the enthalpy of the substance dissolved in the solution to a defined concentration minus the enthalpy of the original substance. The solvent for the dissolution process must be such that the mass of solid dissolves within a time frame that matches the response time of the calorimeter, as discussed below. The enthalpy of solution is proportional to the amount of solid being dissolved. This amount may be defined as 1 mol for molar enthalpy or as 1 g for specific enthalpy. If the substance possesses adequate purity (as determined by the degree of accuracy required) and if its molecular mass is known, the molar enthalpy is preferred, otherwise the specific enthalpy must be used. The enthalpy of solution is weakly dependent on both the temperature, which is usually 25.0 °C, and the final concentration of the dissolved solute.
It is usually preferred to express the crystallinity, Pc, of a substance on a percentage scale. This procedure requires 2 reference standards, namely a highly crystalline sample assuming 100 per cent crystallinity and having a measured enthalpy of solution of 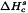 , and an amorphous sample assuming 0 per cent crystallinity and having a measured enthalpy of solution of
, and an amorphous sample assuming 0 per cent crystallinity and having a measured enthalpy of solution of 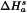 . From these values and from the measured enthalpy of solution,
. From these values and from the measured enthalpy of solution, 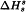 , of the solid under study, the percentage crystallinity of the solid, Pc, may be calculated as follows:
, of the solid under study, the percentage crystallinity of the solid, Pc, may be calculated as follows:
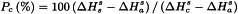
Clearly, crystallinity expressed on a percentage scale depends on 3 measured values and the enthalpies of solution may be replaced by other corresponding physical quantities that depend on crystallinity. The value of the percentage crystallinity of a sample, however, depends not only on the nature and method of preparation of the 2 reference standards, but also on the choice of the physical quantity that is measured.
The enthalpy of solution is measured either by an isoperibol (constant perimeter, i.e. jacket) solution calorimeter or by an isothermal (constant temperature) solution calorimeter. Typically, at least 3 measurements are made with each sample. The mean of these values is then calculated. The exact requirements will depend upon the equipment capability and degree of accuracy needed.
Isoperibol Solution Calorimetry
In the isoperibol solution calorimeter, the heat change during the solution process causes a corresponding change in temperature of the solvent-solute system (i.e. solution). This temperature change is measured by a temperature sensor, which is wired to an electrical circuit that records an electrical signal corresponding to the temperature change. Typically, this temperature change in an electronic form is measured at precisely defined time intervals to produce temperature-time data that are collected, analysed by a computer, and then plotted. A blank run without addition of the solid solute to the solvent normally shows no discernible change in the slope of the temperature-time plot.
For isoperibol solution calorimeters, response is fairly rapid, but corrections must be made for any heat losses to or heat gains from the bath. Therefore, isoperibol solution calorimeters are more advantageous than isothermal solution calorimeters when the solution process is relatively fast. For all measurements of enthalpy of solution using isoperibol solution calorimeters, the choice of solvent is critical. The nature and mass of the solvent and the mass of sample allow the total heat change, corresponding to total dissolution of the solid, to proceed to completion within 5 min under vigorous stirring at a constant rotational speed within the range of 400-600 r/min.
The effective heat capacity of the calorimeter cell and its contents is determined for every calorimeter run. This determination is accomplished by electrical heating of the contents of the calorimeter cell. The effective heat capacity is determined according to 1 of 2 protocols: either by making 1 determination after ampoule breakage or by making 1 determination before and a 2nd determination after ampoule breakage and then averaging the 2 results. The accuracy and reliability of the electrical heating are established by the accuracy and reliability of the aforementioned chemical calibrations.
Isothermal Solution Calorimetry
In the isothermal (constant temperature) solution calorimeter, the heat change during the solution process is compensated for by an equal but opposite energy change, such that the temperature of the solvent-solute system (i.e. solution) remains essentially constant. This equal but opposite energy change is measured and, when its sign is reversed, provides the enthalpy of solution. For isothermal calorimeters, response is relatively slow, but the compensation process eliminates the effects of heat losses to or heat gains from the bath. Therefore, isothermal solution calorimeters are more advantageous than isoperibol solution calorimeters when the solution process is relatively slow.
SOLUTION CALORIMETER CALIBRATION
To ensure the accuracy of the calorimeter, chemical calibrations must be performed on a regular basis. For an endothermic solution process, the calibration of the calorimeter is checked by measuring the heat absorbed during the dissolution of potassium chloride in distilled water at 298.15 K (25.0 °C). The established enthalpy change in this endothermic process is 235.5 J/g (17.56 kJ/mol). For an exothermic solution process, the calorimeter is checked by measuring the heat evolved during the dissolution of 5 g per litre of tromethamine [tris(hydroxymethyl)aminomethane, THAM] in a 0.1 mol/L aqueous hydrochloric acid solution at 298.15 K (25.0 °C). The established heat for the aforementioned process is -246.0 J/g (-29.80 kJ/mol).
SAMPLE HANDLING
The chemical and physical stability of solids may decrease with decreasing crystallinity. In particular, solids of low crystallinity, especially amorphous solids, tend to sorb water vapour from the atmosphere, leading to crystallisation and a corresponding gain in crystallinity. For these reasons, anhydrous samples whose crystallinity is to be determined must be stored at zero humidity or below critical humidity levels in sealed chambers containing a desiccant, preferably containing an indicator of effectiveness. If crystallinity-humidity studies are to be carried out, the sample is stored in a sealed chamber containing a saturated salt solution to provide a defined relative humidity.