Appendix XVII U. Crystallinity
INTRODUCTION - THE CONCEPT OF CRYSTALLINITY
Most organic and inorganic compounds of pharmaceutical relevance exist as a solid material, which can be characterised by a structure located between a perfectly ordered crystal and an amorphous material.
Real crystals lie somewhere between an ideal crystal state and the amorphous state. The position of a crystal on a scale bounded by these 2 extremes is termed its crystallinity.
A perfectly ordered crystal is an ideal state that is seldom, if ever, achieved. The structural units of a crystal, termed unit cells, are repeated regularly and indefinitely in 3 dimensions in space. The unit cell has a definite orientation and shape defined by the translational vectors a, b and c, and the angles α, β and γ, and hence has a definite volume, V, that contains the atoms and molecules necessary for forming the crystal. A crystalline system is defined by 3 long-range order symmetry operators (translational, orientational and conformational); the various mesophases (liquid crystals, crystals and plastic crystals) have 1 or 2 of the long-range symmetry operators and the ideal amorphous state is defined by the absence of all 3 operators.
Each crystal can be classified as a member of one of 7 possible crystal systems that are defined by the relationships between the individual dimensions a, b and c and between the individual angles α, β and γ of the unit cell. The structure of a given crystal may be classified according to one of the 7 crystal systems, to one of the 14 Bravais lattices and to one of the 230 space groups. All the 230 possible space groups, their symmetries and the symmetries of their diffraction patterns are compiled in the International Tables for Crystallography.
Many substances are capable of crystallising in more than one type of crystal lattice, which is known as polymorphism. The occurrence of polymorphism is a common phenomenon among organic molecules, giving rise to different physico-chemical properties. Crystalline polymorphs have the same chemical composition but different internal crystal structures and, therefore, possess different physico-chemical properties. The different crystal structures in polymorphs are due to different atomic packing arrangements and/or different conformations of the molecules (see chapter 5.9. Polymorphism).
The other extreme of a crystal state is the ideal or true amorphous state, where all long-range order is lost. For most organic systems certain short-range order remains, but this is not expected to extend over distances much larger than nearest neighbour (NN) or next nearest neighbour (NNN) interactions, which are typically less than 2-2.5 nm for small organic molecules.
Amorphous material is characterised by the absence of distinct reflections in the X-ray powder diffraction (XRPD) pattern (2.9.33).
The crystallinity of a real powder can be considered by 2 models of crystallinity. In the 1-state model all particles will be of the same crystallinity whereas in the 2-state model each particle can be either crystalline or amorphous, such that the actual crystallinity of the powder is the weighted average of these 2 extreme crystallinities. Such a powder is obtained when pure crystalline and amorphous phases are physically mixed. In reality, a powder probably contains particles with different degrees of crystallinity, just as it may contain particles with different sizes and shapes.
The extent of disorder in a crystalline solid can affect many physico-chemical properties of substances for pharmaceutical use. Because of the great relevance of these properties, it is important to be able to assess the extent of disorder or the crystallinity of a solid by a suitable quantitative method.
METHODS FOR MONITORING AND DETERMINinG CRYSTALLINITY
Various methods are available for determining the crystallinity of a solid. Many techniques cannot detect or quantify these properties independently; for this reason, it is useful to combine several of the methods described below. Such methods often do not give accurate results and limits of quantitation are usually much greater than those for chemical impurities. In addition, certain assumptions have to be made about the relationship between standards used for calibration, which are typically mixtures of crystalline and amorphous particles (2-state model), and the samples to be analysed that are likely to have at least a small component of material exhibiting 1-state model behaviour. Finally, the lack of well-defined standards for 100 per cent crystalline or 100 per cent amorphous material complicates the validation of such methods. As explained above, it is obvious that different amorphous or non-crystalline phases exist and even co-exist in a solid powder. These different non-crystalline forms of a solid can give different responses depending on the techniques used for determining the degree of crystallinity.
X-ray powder diffraction (2.9.33)
XRPD is still the most commonly used method for determining the degree of crystallinity, although this method suffers from some limitations due to peak broadening, amorphous halo and preferred orientation, which make interpretation and quantitation difficult.
XRPD alone is often insufficient to distinguish between the different non-crystalline phases. The X-ray diffraction pattern of a purely amorphous and nanocrystalline phase is characteristic of a broad diffuse halo. In-depth analysis of the X-ray diffraction patterns will show that the diffuse halo in the pattern of nanocrystalline material shows some correlation to the pattern of the parent crystalline phase, while in the case of a pure amorphous phase such a correlation does not exist. Additional techniques may be required to establish the true nature of X-ray amorphous materials.
Thermal analysis
Thermal analysis (2.2.34) of crystalline materials exhibits a melting transition that is often accompanied by decomposition or evaporation of solvents. In the case of true amorphous materials, thermal analysis reveals a glass transition, whereas only a melt would be expected for a nanocrystalline material.
Microcalorimetry (2.2.61)
It is a highly sensitive technique which allows the determination of the rate and extent of chemical reactions, changes of phase or changes of structure. Amorphous parts of a substance can recrystallise by subjecting the sample to higher relative humidity or an atmosphere containing organic vapour. The measurement of the heat of recrystallisation enables the amorphous content to be determined from the enthalpy of recrystallisation. By relating the output from the microcalorimeter for a sample to that obtained for an amorphous standard, it is possible to quantify the amorphous content of the sample. The range of amorphous content covered by this method depends on the individual substance to be tested; in favourable cases limits of detection below 1 per cent can be reached.
Solution calorimetry (2.2.61)
Solution calorimetry provides a means of determining enthalpy of solution for a solid substance. The crystallinity of the solid sample to be examined is given by the enthalpy of solution of the solid sample 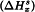 minus the enthalpy of solution of the chosen reference standard of the same substance
minus the enthalpy of solution of the chosen reference standard of the same substance 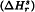 when determined under the same conditions. Because the reference standard is usually chosen for its perceived high crystallinity, its enthalpy of solution is usually algebraically greater (more endothermic or less exothermic) than that of the solid sample to be examined in the same solvent. Consequently, the crystallinity determined is a negative quantity with the SI units kJ/mol or J/g (J/kg is avoided because of its unwieldiness and potential for error). The preference for a negative value with respect to a highly crystalline reference standard recognises the fact that most samples have a lower crystallinity than this reference standard.
when determined under the same conditions. Because the reference standard is usually chosen for its perceived high crystallinity, its enthalpy of solution is usually algebraically greater (more endothermic or less exothermic) than that of the solid sample to be examined in the same solvent. Consequently, the crystallinity determined is a negative quantity with the SI units kJ/mol or J/g (J/kg is avoided because of its unwieldiness and potential for error). The preference for a negative value with respect to a highly crystalline reference standard recognises the fact that most samples have a lower crystallinity than this reference standard.
Near-infrared (NIR) spectroscopy
Near-infrared (NIR) spectroscopy (2.2.40) is another technique used to measure the degree of crystallinity, and has also been proven to be useful in studies of polymorphism. The NIR spectrum of a sample contains both physical and chemical information. Being non-invasive, non-destructive and operable at room temperature, the method is a valuable tool to assess changes in the amorphous and crystalline state.
Infrared absorption spectrophotometry and Raman spectrometry
Infrared absorption spectrophotometry (2.2.24) and Raman spectrometry (2.2.48) are other techniques used to measure the degree of crystallinity, and have also been proven to be useful in studies of polymorphism. The IR spectrum and Raman spectrum of a sample contain both physical and chemical information.
Solid-state NMR
Solid-state nuclear magnetic resonance spectrometry (ss NMR) (2.2.33) can be used to provide information about polymorphism and related relative molecular conformations. However, some caution has to be exercised in the interpretation of results, since it is not always simple to distinguish between samples that comprise a mixture of different physical forms (2-state model) and those that comprise crystals having disorder with exchange that is slow on the NMR timescale. Similarly, samples that contain defects arising from different molecular conformations or slightly different packing arrangements (1-state model) may show additional signals in the spectra. Solid-state NMR may be quite sensitive to this, even if lattice parameters are hardly affected and, consequently, little or no change is observed by XRPD. It is evident that the crystallinity of substances for pharmaceutical use can be complex, and both crystalline defects and amorphous material may co-exist.
Optical microscopy
A method to detect whether or not particles are crystalline is to use a polarising microscope (2.9.37), where particles show birefringence and extinction positions when the microscope stage is revolved.