Ranitidine Tablets
Action and use
Histamine H2 receptor antagonist; treatment of peptic ulcer disease.
Definition
Ranitidine Tablets contain Ranitidine Hydrochloride. They are coated.
Content of ranitidine, C13H22N4O3S
95.0 to 105.0% of the stated amount.
Identification
Shake a quantity of the powdered tablets containing the equivalent of 25 mg of ranitidine with 5 mL of methanol for 5 minutes, filter and evaporate the filtrate to dryness. Add 1 mL of petroleum spirit (boiling range, 60° to 80°) to the residue, scratch the side of the vessel to induce crystallisation, evaporate to dryness and dry the residue at 60° for 10 minutes. The infrared absorption spectrum of the dried residue, Appendix II A, is concordant with the reference spectrum of ranitidine hydrochloride (RS 311).
Tests
Dissolution
Comply with the requirements for Monographs of the British Pharmacopoeia in the dissolution test for tablets and capsules, Appendix XII B1.
Calculate the total content of C13H22N4O3S in the medium from the absorbances obtained and using the declared content of C13H22N4O3S in ranitidine hydrochloride BPCRS.
Related substances
Carry out the method for liquid chromatography, Appendix III D, using the following solutions.
Mobile phase A
Mobile phase B
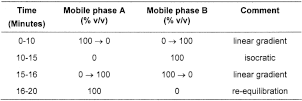
When the chromatograms are recorded under the prescribed conditions the retention times relative to ranitidine (retention time about 7 minutes) are; impurity H, about 0.1; impurity D, about 0.8; impurity J, about 0.9 and impurity A, about 1.7.
The test is not valid unless, in the chromatogram obtained with solution (4), the resolution between the impurity J and ranitidine is at least 1.5.
Identify any peak due to impurity A and impurity J in the chromatogram obtained with solution (1), using the chromatogram obtained with solutions (3) and (4) respectively. Multiply the area of the peak due to impurity J by a correction factor of 2.
In the chromatogram obtained with solution (1):
the area of any peak corresponding to impurity A is not greater than 2.5 times the area of the principal peak in the chromatogram obtained with solution (2) (0.5%);
the area of any other secondary peak is not greater than the area of the principal peak in the chromatogram obtained with solution (2) (0.2%);
the sum of the areas of any secondary peaks is not greater than 5 times the area of the principal peak in the chromatogram obtained with solution (2) (1.0%).
Disregard any peak with an area less than half the area of the principal peak in solution (2) (0.1%).
Assay
Carry out the method for liquid chromatography, Appendix III D, using the following solutions.
The assay is not valid unless the peak due to ranitidine in the chromatogram obtained with solution (3) shows baseline separation from the peak due to dimethyl{5-[2-(1-methylamino-2-nitrovinylamino)ethylsulfinylmethyl] furfuryl}amine.
Calculate the content of C13H22N4O3S in the tablets using the declared content of C13H22N4O3S in ranitidine hydrochloride BPCRS.
Storage
Ranitidine Tablets should be protected from light.
Labelling
The quantity of active ingredient is stated in terms of the equivalent amount of ranitidine.
IMPURITIES
The impurities limited by the requirements of this monograph include those listed under Ranitidine Hydrochloride.