Appendix V R. Detection and Measurement of Radioactivity
INTRODUCTION
Within the context of the European Pharmacopoeia, the term ‘radioactivity’ is used both to describe the phenomenon of radioactive decay and to express the physical quantity of this phenomenon. In the monographs on radiopharmaceutical preparations, the detection and measurement of radioactivity are performed for different purposes: verification of the characters, identification, determination of radionuclidic and radiochemical purity, as well as determination of the radioactivity in a substance (assay).
Under these assumptions, the measurement can be qualitative, quantitative or both, depending whether it is directed to the identification of the radionuclide or the determination of its activity (rate of decay) or both of them.
Radioactive sources can produce various types of emissions, such as alpha particles, electrons, positrons, gamma- and X-rays, according to the radionuclidic composition.
Each radionuclide yields characteristic emissions, with specific energies and relative intensities. Such radiations can be detected as a result of their ionising properties in an ionisation chamber but without further characterisation; when they are detected and analysed using a spectrometer, an energy spectrum is obtained. A detailed spectrum analysis is typically used to identify radionuclides present in a sample. Spectrometry can also be used for quantitative determination of the radioactivity in sources made of a single radionuclide or radionuclide mixtures or of the individual radionuclides present.
A measurement of radioactivity is generally performed by counting the number of detected decay events (emissions). Therefore, the geometry of the sample during the measurement of radioactivity and the acquisition time strongly influence the result. In general, the measurement geometry must correspond to a calibrated geometry and the acquisition time must be long enough to reach sufficient counting statistics.
A measurement of radioactivity can be done in a stand-alone mode (e.g. using an ionisation chamber or a spectrometer) or in combination with a separation technique (e.g. radiochromatography) to account for relative contributions from different radioactive chemical species that may be present in a mixture.
MEASUREMENT OF RADIOACTIVITY
A direct determination of the radioactivity of a given sample, in becquerel (Bq), may be carried out if the decay scheme of the radionuclide is known, but in practice many corrections are required to obtain accurate results. For this reason, it is possible to carry out the measurement with the aid of a primary standard source or by using measuring instruments such as an ionisation chamber or a spectrometer calibrated using suitable standards for the particular radionuclides.
A spectrometer is used when measuring the radioactivity of radionuclides in a mixture, each radionuclide being identified by its emissions and their characteristic energies.
All measurements of radioactivity must be corrected for dead-time losses and by subtracting the background signal due to radiation in the environment and to spurious signals generated in the equipment itself.
The radioactivity of a preparation is stated at a given date. If the half-life of the radionuclide is less than 70 days, the time is also indicated. This statement of the radioactive content must be made with reference to a specified time zone. The radioactivity at other times may be calculated from the exponential decay equation or from tables.
In general, a correct measurement of radioactivity requires that consideration is given to some or all of the following:
Dead-time losses
Due to the finite resolving time (dead time) of the detector and its associated electronic equipment, it may be necessary to correct for losses by coincidence. The resolving time of a counter is the minimum time interval required by the counter to resolve 2 single pulses. Incident radiation events at shorter intervals may not be detected or may be detected as a single event with the summed energy. These losses are sometimes referred to as ‘dead-time losses’. For a counting system with a fixed dead time τ following each count, the true count rate, per second, is calculated using the following expression:
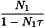
| N1 | = | the observed count rate, per second; |
| τ | = | the dead time, in seconds. |
With some equipment this correction is made automatically. Corrections for losses by coincidence must be made before the correction for background radiation.
Correction for decay during measurement
If the time period of an individual measurement, tm, is not negligibly short compared with the half-life of the radionuclide, T1/2, the decay during this measurement time must be taken into account. For example, there is a 5 per cent cumulative loss of counts due to decay during a counting period that is 15 per cent of the half-life of the radionuclide.
After having corrected the instrument reading (count rate, ionisation current, etc.) for background signals and, if necessary, for losses due to electronic effects, the instrument reading corrected to the beginning of the individual measurement is calculated using the following expression:
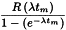
| R | = | instrument reading before decay correction, but already corrected for background signal, etc.; |
| λ | = | radionuclide decay constant (ln 2/T½); |
| e | = | base of natural logarithm; |
| tm | = | measurement duration. |
Statistics of radioactivity measurement
The results of determinations of radioactivity show variations that derive mainly from the random nature of nuclear transformations. Counting for any finite time can yield only an estimate of the true rate of nuclear transformations. A sufficient number of counts must be registered in order to compensate for variations in the number of transformations per time. In the case of measurement of radioactivity, the standard deviation of the recorded counts is the square root of the counts, so at least 10 000 counts are necessary to obtain a relative standard deviation of not more than 1 per cent.
Linearity
The linearity of an instrument is the range of radioactivity for a particular radionuclide over which its efficiency remains constant.
The linear range of a radioactivity measurement assembly can be determined by repeatedly counting a radioactive sample in a fixed geometry as it decays from an activity level that is above the linear range. After correction for the background signal, the natural logarithm of the count rate data is plotted against the elapsed time after the first measurement (Figure 2.2.66.-1).
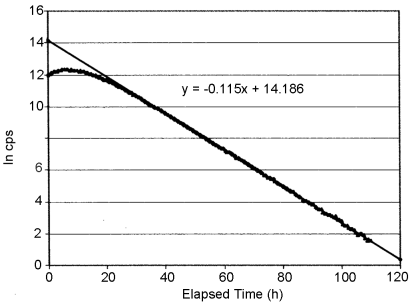
Linear regression analysis of the central, linear portion of the data set yields a slope which is the decay constant λ, which has a characteristic value for each radionuclide:
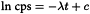
c represents the natural logarithm of the count rate at t = 0 of a perfectly linear instrument.
The resulting regression equation is used to calculate the theoretical count rate at each time that the actual data were recorded. Where the deviation of the measured count rate from the theoretical count rate is unacceptably high, the linear range of the measuring equipment has been exceeded.
Alternatively, a series of dilutions can be made of a radioactive solution of known radioactivity concentration. Equal volumes of each of the dilutions are then counted using standardised geometry and counter settings. The ratio of the count rate for each sample (after correction for background signals and decay) to the calculated radioactivity of the respective sample in Bq is the counting efficiency. The range over which this ratio is constant is the useable range of the measuring equipment for the radionuclide concerned.
The limit of detection and the limit of quantification for equipment and procedures used for radioactivity measurement must be established before their routine use.
Limit of detection
The limit of detection (LOD) of an individual procedure is the lowest amount of radioactivity in a sample that can be detected but not necessarily quantified as an exact value. In practical terms this requires an estimate of the background signal and its standard deviation. The LOD is usually considered to be 3 times the standard deviation of the background signal.
Limit of quantification
The limit of quantification (LOQ) of an individual procedure is the lowest amount of radioactivity in a sample that can be quantitatively determined with suitable precision and accuracy. The LOQ is used particularly for the determination of impurities and/or degradation products. In practical terms the LOQ is usually considered to be 10 times the standard deviation of the background signal.
MEASUREMENT OF RADIOACTIVITY USING IONISATION CHAMBERS
Apparatus
Ionisation chambers (including dose calibrators) are the most common equipment for the measurement of radioactivity in the practice of radiopharmacy. It generally can measure activities from a few tens of kBq to hundreds of GBq. It usually comprises a sealed well-type ionisation chamber and built-in electronics to convert the detector signal to a measure of radioactivity.
The chamber is filled with a gas across which an electrical field is applied. When the gas is ionised by the radiation emitted by the source, the resulting ionisation current is measured and related to the radioactivity present in the ionisation chamber. The ionisation current is influenced by the applied voltage, the energy and the intensity of the radiation and the nature and pressure of the gas. The instrument settings (calibration factor) may be adjusted to keep a direct relationship between the ionisation produced by the radiation of a specific radionuclide and the radioactivity value obtained for each measurement geometry.
As an ionisation chamber measures only the current resulting from the overall ionisation produced within the chamber, it cannot discriminate between the emissions of different radionuclides.
For an accurate measurement of the radioactivity of a specific radionuclide, the measurement must be corrected for the contributions to the ionisation current caused by radionuclidic impurities present in the preparation.
The activity levels to be measured are limited by saturation considerations, the range of the amplifier and the design of the chamber itself. The linearity range of the ionisation chamber is established as described above under Linearity.
The ionisation chamber must be shielded to minimise background signals to an acceptable level.
Method
The sample is positioned inside the well of the ionisation chamber at a given position, using a holder. After putting the sample in the ionisation chamber, the activity reading is made once the response is stable. Measuring the sample under exactly the same geometrical conditions as the calibration source will yield the most accurate results. If necessary, dilute the preparation to be measured to the same volume as that of the calibration source.
Calibration The ionisation chamber is calibrated taking into account the shape, dimensions, material of the container, volume and composition of the solution, the position within the chamber and the radionuclide being measured. Limits for uncertainty in calibration can be found in national and international regulations.
Calibrate the ionisation chamber at least once a year, by using sources of radionuclides traceable to national or international standards in the appropriate containers (vial, syringe) with regard to geometry. Establish and implement subsidiary correction factors to take account of the differing configurations of the radionuclides to be measured. Perform a linearity check of the instrument’s response over the complete range of energies and activities for which the equipment is used.
For each setting and before each use (minimum once on each day of use) perform a constancy check of the ionisation chamber using standard sources of radionuclides with long half-lives to verify its calibrated state. A check with a reference source, such as caesium-137, must be performed on each day of use to verify that the ionisation chamber is still in its calibrated state.
MEASUREMENT OF RADIOACTIVITY USING SOLID-STATE DETECTORS
Solid-state detectors include scintillating plastic fluors and crystals, and semiconductors. Further to their application in spectrometry (see section Spectrometry), solid-state detectors can be used for the measurement of radioactivity. In particular, due to their high sensitivity, plastic and crystal scintillation detectors are used in counting low levels of radioactivity. Dead-time losses must be carefully considered with these types of detectors. Semiconductor detectors are used when a higher energy discrimination is required, for example in mixtures of radionuclides or when there are potential radionuclidic impurities with emissions of similar energy.
Apparatus
The equipment consists of a shielded detector comprising a plastic or crystal scintillator coupled to a photomultiplier, or a semiconductor, which are connected to an amplifier and counting electronics. The system may have an adjustable energy window, used for selecting a counting region of the radionuclide energy spectrum that may be adjusted by the operator.
Instruments have different properties of energy resolution and detection efficiency depending on the type of detector and its volume and geometry. Lower efficiency requires a longer counting time.
Samples to be measured may be placed in front of the detector or into the well of a well-type detector. Measuring chambers may be enclosed in the detector shielding and single samples may be introduced using lids or other positioning systems to ensure correct measurement geometry.
A scintillation detector can be used for dynamic radioactivity measurement when, for example, the eluate of a liquid chromatograph is directed over or through a detector, see section on Detection and measurement of radioactivity in combination with a separation technique.
Method
Ensure that the sample radioactivity gives a counting rate in the linearity range of the equipment. The measurement is started after any shielding is in place or the well cover is replaced and the counting time is selected to reach sufficient counts for a statistically significant value.
Calibration The detector has to be calibrated by measuring its efficiency using a source of the radionuclide in question traceable to national or international standards. Calibration in terms of efficiency uses sources such as caesium-137, cobalt-60, barium-133 and others covering the desired energy range.
MEASUREMENT OF RADIOACTIVITY USING LIQUID SCINTILLATION DETECTORS
Liquid scintillation counting is commonly used for beta-particle emitting samples, but is also used for alpha-particle emitting samples. For the principles of the detection of radioactivity using liquid scintillation detectors see under Beta-particle spectrometry below.
Calibration In order to take into account the loss of counting efficiency due to quenching, the liquid scintillation counter may make use of an external source, typically barium-133 or europium-152, which is brought close to the sample vial to release Compton electrons. The shape of the resulting spectrum is analysed automatically to compute a quench-indicating parameter. This parameter can then be related to the counting efficiency measuring sources of known activity at a determined level of quenching agent. The obtained quench curve allows the determination of the activity of an unknown sample knowing the count rate and the value of the quenching parameter.
DETERMINATION OF HALF-LIFE
The half-life is a characteristic of the radionuclide that may be used for its identification. The half-life is calculated by measuring the variation of radioactivity of a sample to be tested as a function of time. Perform the measurements in the linearity range of a calibrated instrument.
Apparatus
Half-life can be measured by using any type of quantitative radioactivity detector provided it is used within a linearity range throughout the range of activities that are present during the measurement and the geometry is not changed during the measurement.
For preparations containing a radionuclide with a short half-life and when stated in a monograph, determination of the approximate half-life contributes to the identification.
Method
Half-life The preparation to be examined is used as such or diluted or dried in a capsule after appropriate dilution. The radioactive sample is prepared in a manner that will avoid loss of material during handling. If it is a liquid (solution), it is contained in a closed flask or a sealed tube. If it is a residue from drying in a capsule, it is protected by a cover consisting of a sheet of adhesive cellulose acetate or of some other material.
The radioactivity of the sample must be high enough to allow measurements over a period corresponding to 3 estimated half-lives but must be, for each measurement, within the linearity range of the equipment. Correction for dead-time losses is applied if necessary.
The same source is measured in the same geometrical conditions and at intervals usually corresponding to at least half of the estimated half-life. Each value is tabulated against the time interval from the initial measurement. To avoid influence of decay during measurement, the counting time is the same for all measurements.
A graph can be drawn with time as the abscissa and the logarithm of the relative instrument reading (e.g. count rate) as the ordinate. The half-life is calculated from the slope of the best linear fit of the measured values against the time corresponding to each measurement.
Approximate half-life For this purpose, not fewer than 3 measurements are made over a period of not less than 1/4 of the estimated half-life.
The sample to be examined and the instrument to be used comply with the indications given above. The data are processed in the same way as above.
SPECTROMETRY
Radionuclides can be identified by their emission spectrum. Each type of emission (i.e. alpha particles, beta particles and electrons, gamma- and X-rays) requires specific equipment to acquire an emission spectrum. Spectrometers must be calibrated in order to work properly and the following sections describe the different equipment and detail the general procedures for a reliable measurement.
Gamma-ray Spectrometry
General principles
In gamma-ray spectrometry using a scintillation detector, absorption of gamma- and X-rays results in production of light, which is converted into an electrical pulse by a photomultiplier. In gamma-ray spectrometry using a semiconductor detector, absorption of gamma- and X-rays results in the immediate production of an electrical pulse.
In both cases the pulse amplitude is proportional to the energy of the absorbed radiation. The most common detectors for gamma- and X-ray spectrometry are thallium-activated sodium iodide (NaI(Tl)) scintillation counters and high-purity germanium (HPGe) semiconductor detectors.
A gamma-ray spectrum can be produced by collecting and analysing a sufficient number of pulses.
Apparatus
A gamma-ray spectrometer usually comprises a shielded measuring chamber where the sample is positioned, a detector, an electronic chain and a multichannel analyser.
The shielding of the chamber must be able to reduce the background signal to a level that allows the registration of a correct gamma-ray spectrum.
The measurement chamber has a movable cover or a drawer to allow the positioning of the sample. A sample holder may be present to ensure reproducible geometry between measurements.
The duration of measurement is related to the radioactivity of the target radionuclide and a long period of acquisition may be required to achieve the necessary counting statistics. Dead-time losses must be carefully considered with this type of detector.
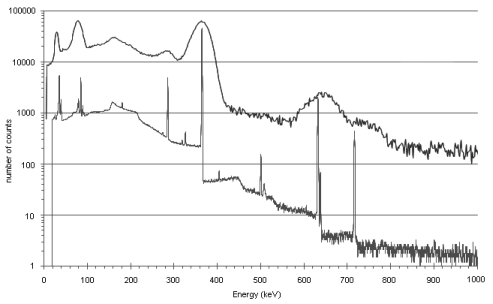
The sensitivity of a NaI(Tl) detector is higher than that of a germanium detector of the same size. In general, peaks in an energy spectrum are identified with an uncertainty depending upon the full width of the peak at its half-maximum height (FWHM). The energy resolution of a solid-state scintillation detector is much poorer than that of a semiconductor detector and hence peaks obtained with a semiconductor detector are much narrower than those obtained with a scintillation detector. Figure 2.2.66.-2 shows a comparison of the spectra obtained from the same source with the 2 types of detector.
The different performances of NaI(Tl) and HPGe detectors may limit their use in some spectrometric analyses.
For the identification of the radionuclide(s) in a preparation and determination of radionuclidic purity, a risk assessment on the process of radionuclide production must assess the potential presence of other radionuclides with photon energies in the same range (± 10 per cent) as that of the radionuclide(s) present in the radiopharmaceutical.
In case radionuclidic impurities can be present that emit gamma- or X-rays with an energy in the same range as that of the photons emitted by the radionuclide in the preparation, a measured peak energy within a maximum interval of ± 2 keV or ± 2 per cent (whichever is the larger) with respect to the nominal peak energy (see 5.7. Table of physical characteristics of radionuclides) is sufficient for peak identification.
In the case where such impurities are not expected to be present, a maximum interval of ± 10 keV or ± 6 per cent (whichever is the larger) with respect to the nominal peak energy is acceptable for peak identification.
Method
Ensure that the counting rate of the sample falls within the linearity range of the equipment. For liquid samples this may be achieved by appropriate dilution; for solid samples, by increasing the source-to-detector distance or by using an attenuating material. Introduce the preparation to be examined in a container into the instrument chamber and record the spectrum after closing the shielding.
Ensure that the container used for quantitative measurements is of the same shape, dimensions, volume and material as that of the calibration standard.
Ensure that the composition of the solution and the position of the container in the measuring chamber is the same for the container for the quantitative measurement as for the calibration standard.
Radionuclide identification Calibrate the spectrometer in relation to energy. Determination of the correspondence of the energy of the peaks detected from the sample to the energies prescribed by a monograph is a valid identification test.
Radionuclidic purity Calibrate the spectrometer in relation to efficiency and energy. Determine the LOQ and resolution of the equipment and ensure that they are in line with the limits of the radionuclides to be determined. Record the spectrum of the preparation.
Identify the radionuclides present in the preparation to be examined and determine their radioactivity with the aid of chapter 5.7. Table of physical characteristics of radionuclides. Because the level of radionuclidic impurities, expressed as a percentage of the total radioactivity, may increase or decrease with time, the measured activity of each impurity must be recalculated to the activity during the period of validity of the preparation. The activities of all radionuclidic impurities need to be summed (taking into account the limit of quantification) and related to the total radioactivity of the preparation.
The sample is placed close to the detector or within a well-type detector. All the events within a pre-set energy range are collected and displayed on a ratemeter as counts per second or accumulated over a pre-set period of time. If there is sufficient difference in photon energies emitted by the radionuclide(s), a sodium iodide detector can be suitable, given its high sensitivity. However, if there is a need to discriminate emissions of similar energy, a HPGe detector or another semiconductor detector is needed.
Calibration Calibration in relation to energy is done by using the peaks of known sources traceable to national or international standards, such as cobalt-57, caesium-137, cobalt-60 and others covering the desired energy range. A calibration in relation to efficiency can be simultaneously obtained, so that not only the energy spectrum but also the activity of the sample and the radionuclide impurities can be further determined. The calibration of efficiency can be performed with a traceable radionuclide source with energy peaks covering the desired range or with the aid of a mixed, traceable radionuclide standard with gamma-ray energies covering the desired range.
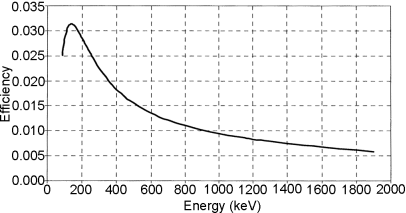
To obtain the efficiency curve, the detector response as a function of the energy has to be measured using each separate sample/detector geometry. For this reason, it is possible to carry out the measurement with the aid of a primary standard source. Primary standards may not be available for radionuclides with a short half-life, e.g. some positron emitters. When measuring, the sample will mostly have to be in a container and set at a defined position in relation to the detector. The sample/detector geometry is then defined by the position of the sample relative to the detector and the characteristics of the container and sample, e.g. shape, volume and density. Figure 2.2.66.-3 shows a typical HPGe detector efficiency curve obtained for a cylindrical container placed on top of the detector.
Beta-particle Spectrometry
In the case of a beta-particle emitter, a beta-particle spectrometer is necessary to determine the energy distribution of the emitted beta particles. It is analogous to a gamma-ray spectrometer, but frequently uses liquid scintillators to convert the energy of the beta particles into detectable light, which can then be analysed. Beta-particle spectrometry is mostly achieved by dissolving or suspending the sample in a liquid scintillation cocktail in transparent or translucent (glass or plastic) containers and subsequent counting of the electrical pulses generated by a photomultiplier from the emitted light. The pulse amplitude is related to the energy of the absorbed radiation. A beta-particle spectrum can be produced by collecting a sufficient number of pulses. The liquid scintillation cocktail is chosen in such a way that counting errors due to quenching, chemoluminescence, phosphorescence, etc., are minimised. Coincidence counting with 2 or more photomultipliers is also used to minimise counts from background radiation, electronics, etc.
To differentiate between alpha- and beta-particle emissions, pulse-shape discrimination is commonly used.
Radionuclide identification Determination of the correspondence of the mean and/or maximum energies in the energy spectrum from the sample to the energies prescribed by a monograph is a valid identification test.
Calibration A common method of energy calibration is to use an unquenched reference sample to determine the maximum energy of the beta particles emitted by the radionuclide of interest.
Alpha-particle Spectrometry
For the identification and assay of alpha-particle emitters, spectrometry using liquid scintillation is mostly used. The principle is explained in the previous section on beta-particle spectrometry.
For the identification and determination of radionuclidic purity of alpha-particle emitters, spectrometry using a silicon-diode semiconductor detector can be used. Using this detector, the absorption of alpha particles results in the immediate production of an electrical pulse. The movement of electron-hole pairs created by the interaction of radiation induces an electrical charge, which is amplified and measured.
The sample preparation is of crucial importance. After a chemical separation of the radionuclide of interest, the sample is electro-deposited on a stainless steel disk in the form of a very thin layer of material to minimise self-absorption. The yield of the whole procedure can be determined experimentally by adding a known amount of a tracer, which will take into account the chemical separation efficiency, the electro-deposition efficiency and the counting efficiency.
For both types of detectors, the pulse amplitude is related to the energy of the absorbed radiation. An alpha-particle spectrum can be produced by collecting a sufficient number of pulses.
Radionuclide identification Determination of the correspondence of the energy of the peaks detected from the sample to the energies prescribed by a monograph is a valid identification test.
Calibration An alpha-particle spectrometer has to be calibrated in relation to energy and efficiency. This is done by using the peaks from known sources covering the desired energy range, such as americium-241 and plutonium-242. Not all alpha particles emitted by the source will produce a count in the system. The probability that an emitted alpha particle will interact with the detector material and produce a count is the efficiency of the detector, which depends on the geometry.
DETECTION AND MEASUREMENT OF RADIOACTIVITY IN COMBINATION WITH A SEPARATION TECHNIQUE
A radioactive preparation may contain the radionuclide in different chemical forms other than the intended one. Therefore it is necessary to separate the different substances containing the radionuclide and determine the percentage of radioactivity due to the radionuclide concerned associated with the stated chemical form and the contribution to the total radioactivity due to the radionuclide concerned coming from other substances. For this purpose, instruments for the detection and measurement of radioactivity are used in combination with a physico-chemical separation technique. In principle, any method of separation may be used.
Monographs for radiopharmaceutical preparations may include the combined use of radioactivity measurement with paper chromatography (2.2.26), thin-layer chromatography (2.2.27), gas chromatography (2.2.28), liquid chromatography (2.2.29), size-exclusion chromatography (2.2.30) or electrophoresis (2.2.31).
In all cases the radioactivity of each analyte is measured after the separation has been achieved using the stated method.
Radioactivity measurement may be performed using detectors mounted in series with other detectors in analytical instruments, such as liquid chromatographs, making in-line detection of analytes, or performed off-line, i.e. after the analytical separation has been completed, by measuring the radioactivity of eluate fractions obtained after liquid chromatographic separation of equal volume or as the distribution of radioactivity on paper chromatography or thin-layer chromatography supports.
In-line detection and measurement of radioactivity in combination with liquid chromatography
Apparatus
Liquid chromatography (see 2.2.29) may be used to separate the principal radioactive substance of a radiopharmaceutical preparation from radiochemical impurities or degradation products. In-line detection is usually obtained by using a scintillation detector connected to a ratemeter and recording device. The scintillating material of the detector is selected on the basis of the emission to be detected, e.g. plastic scintillator for beta-particle emissions or scintillation crystals for gamma- and X-ray radiations. The addition of a liquid scintillation cocktail before the eluate reaches the in-line radioactivity detector may also be used in the case of beta-particle-emitting radionuclides.
The simultaneous use of a radioactivity detector and other detectors (UV, refractive index, conductimetric, etc.) connected in series may be used to identify the substance, e.g. in relation to the retention time of a known standard, to determine the amount of the substance using a suitable reference standard and to measure the radioactivity associated with such a substance. When different detectors are coupled in series, correct the experimentally obtained retention times for the delay in time between the detectors.
In liquid chromatography some radiochemical impurities, such as colloidal impurities, may be retained on the column. In such cases a separate method is required for the determination of the content of the retained radiochemical impurities and the calculation formula for the expression of the total radiochemical purity takes into account the relative amount of the retained radiochemical impurities.
One possibility to evaluate such retention problems during method validation is to evaluate the radioactivity recovery from the column by measuring the total radioactivity recovered from the chromatographic equipment with and without the column.
Method
The sample is diluted if necessary and then applied to the column in the prescribed volume and conditions. In this respect it is important to demonstrate the LOD and LOQ, and the linearity of the detector throughout the range of activities to be measured.
Flow-through detector A portion of the tubing where the eluate containing the radioactive species is flowing is placed in front of or within the detector. Counting efficiency may be increased using a longer portion of the tube (e.g. making multiple turns in front of or within the detector); however, this will reduce the ability of the system to separate 2 closely eluting peaks of radioactivity.
When the radiochemical purity test prescribes determination of the total radiochemical impurities or there is a quantitative determination of an individual impurity, it is important to choose an appropriate threshold setting and appropriate conditions for integration of the peak areas. In such tests the disregard limit, i.e. the limit at or below which a peak is disregarded, is dependent on the method and is related to the limit of detection and limit of quantification. Thus, the threshold setting of the data collection system corresponds to at least half of the disregard limit.
Record the signal of the detectors as a function of time.
Identification of peaks in the radiometric signal (radiochromatogram) is made on the basis of the retention time of the analytes. The profile from other detectors may be used for this purpose.
Quantification of the different components of chromatogram and radiochromatogram profiles is made on the basis of peak areas. Peak areas are usually obtained by direct integration of the detector signal using commercially available software.
Off-line detection and measurement of radioactivity
Liquid chromatography (2.2.29). Provided the retention times of the various radiochemical species are reproducibly consistent, an alternative method of radioactivity quantification is to collect the liquid chromatography effluent in a series of timed samples (fractions) for off-line analysis for radioactivity content. The radioactivity in the fractions corresponding to the peaks can be expressed as a percentage of the total of the radioactivity in all fractions, taking into account the limit of quantification.
Method
The sample is applied on the column in the prescribed volume and conditions. Fractions are collected at the end of the chromatographic line.
The volume between the detector used to identify the retention time of the peaks and the collection point is measured and a delay factor is calculated on the basis of the effluent flow rate and applied to each peak to estimate the time of elution of the peak at the point of collection. The fractions are collected on the basis of a fixed time interval or at the time of appearance estimated from the delay time so that any relevant peak is collected in one or more fractions.
The radioactivity of each fraction is counted using a calibrated instrument such as a dose calibrator or a scintillation detector, taking into account the limit of quantification and the linearity.
An elution profile is obtained tabulating the counts per fraction against the elution time or volume. The activity of fractions belonging to the same peak may be summed and the relative percentage calculated to define radiochemical purity.
Thin-layer chromatography (2.2.27) and paper chromatography (2.2.26). Provided a thin-layer chromatography or a paper chromatography analytical method has been validated for the separation of components of a radioactive preparation, the number and relative intensities of the separated spots can be detected and measured using a radioactivity detector that can relate the radioactivity to a specific position on the chromatographic support.
The positions of the spots (peaks) may permit chemical identification by comparison with solutions of the same chemical substances (non-radioactive), using a suitable detection method.
Apparatus
Scanning device The apparatus generally comprises a radioactivity detector, such as a position-sensitive proportional counter or a collimated scintillation detector placed at a fixed distance from a scanning platform where the chromatographic support to be scanned is positioned.
The radioactivity of the sample applied to the chromatography support must result in a counting rate in the linearity range of the equipment and the sample may be diluted if necessary. The area to be scanned is positioned at the reference position so that the desired lane is aligned with the detector scanning trip. Adjust the scanning time to allow enough counting time during the run.
The detector or the platform may be moved in-plane, along the x-axis or the y-axis, so that the entire surface can be scanned during a single run.
The detector is connected to a suitable counting device, so that the radioactivity revealed can be measured quantitatively and the count rate related spatially to the surface scanned.
The radioactivity is automatically reported against the development distance and the profile describes peaks having an area proportional to the number of counts per unit of distance.
Radioactivity counter In the case where a maximum of only 3 radiochemical components needs to be identified and they are fully separated, the support can be cut into equal strips, each having a size not more than half the length of the support corresponding to the difference between the retardation factors of the 2 closest spots. Each single strip is numbered starting from the origin side and counted separately. Alternatively, for well-characterised systems the support may be cut into 2 or more unequal portions, folded if necessary to approximately equal geometry before counting. An ionisation chamber or a scintillation counter can be used for this purpose, provided they are used within the instrument’s linearity range and above its LOQ.
Autoradiography This may also be used to acquire an image of the radioactivity distribution on the chromatographic support. In this case, the response of the system used for the acquisition of the image, such as a phosphor imager or a photographic film, must be shown to be linear with respect to the radioactivity in the chromatogram. Otherwise the system must be pre-calibrated or exposed at the same time to a series of reference radioactive sources, obtained by dilution from a calibrated standard solution, covering the expected radioactivity range that may be present on the support.
Method
Deposit the required amount of sample at the origin of the chromatographic support, with drying if necessary to avoid spreading of the spot. Develop the chromatogram according to the prescribed method. A carrier may be added when prescribed in a particular monograph.
In paper and thin-layer chromatography, it is preferable not to dilute the preparation to be examined but it is important to avoid depositing such a quantity of radioactivity that counting losses by coincidence (dead-time losses) occur during measurement of the radioactivity.
After development, the support is dried and the positions of the radioactive areas are detected by measurement of radioactivity over the length of the chromatogram, using a suitable collimated counter, by autoradiography, or by cutting the strips into portions and counting each portion.
Radioactivity may be measured by integration using an automatic-plotting instrument or a digital counter.
The ratios of the areas under the peaks give the ratios of the percentages of radioactivity due to the respective radiochemical substances.
When the strips are cut into portions, the ratios of the quantities of radioactivity measured give the ratio of percentages of radioactivity due to the respective radiochemical species.
Calibration It is important to demonstrate the limits of detection and quantification, and the linearity of the detector throughout the range of activities to be measured and in all positions on the support of the chromatographic system. This may be done by applying samples covering a range of activities from 0.1 per cent to 100 per cent of the expected range. Prepare the samples by dilution and apply equal volumes of each, with drying if necessary. After examining the radioactivity profile using the equipment’s standard settings, the peak areas are integrated for comparison with the calculated amount of radioactivity applied to each spot. Verify that the response of the detector over the complete length and width of the detector path is the same, as the response may vary with the detector position.
The peak-resolving power is influenced by the size of the spot, the total radioactivity of the radionuclide and the detector equipment. It can be checked by applying 5 µL spots separated by distances increasing from 4 mm to 20 mm in 2 mm increments. The approximate resolution of the detection system can be determined from the radioactivity profile as the distance between the 2 spots where the baseline is only just clearly separated.