Appendix XIV K. Immunological Products
1. Assay of diphtheria vaccine (adsorbed)
The potency of diphtheria vaccine is determined by administration of the vaccine to guinea-pigs followed either by challenge with diphtheria toxin (method A or B) or by determination of the titre of antibodies against diphtheria toxin or toxoid in the serum of guinea-pigs (method C). In both cases, the potency of the vaccine is calculated by comparison with a reference preparation, calibrated in International Units.
The International Unit is the activity contained in a stated amount of the International Standard, which consists of a quantity of diphtheria toxoid adsorbed on aluminium hydroxide. The equivalence in International Units of the International Standard is stated by the World Health Organization (WHO).
Diphtheria vaccine (adsorbed) BRP is suitable for use as a reference preparation.
The method chosen for the assay of diphtheria vaccine (adsorbed) depends on the intended purpose. Method A or B is used:
Method A or B may also be used for the routine assay of batches of vaccine, but in the interests of animal welfare, method C is used wherever possible.
Method C may be used, except as specified under 1 and 2 above, after verification of the suitability of the method for the product. For this purpose, a suitable number of batches (usually 3) are assayed by method C and method A or B. Where different vaccines (monovalent or combinations) are prepared from diphtheria toxoid of the same origin, and with comparable levels (expressed in Lf/mL) of the same diphtheria toxoid, suitability demonstrated for the combination with the highest number of components can be assumed to be valid for combinations with fewer components and for monovalent vaccines. Any combinations containing a whole-cell pertussis component or containing haemophilus type b conjugate vaccine with diphtheria toxoid or CRM 197 diphtheria protein as carrier in the same vial must always be assessed separately.
For combinations containing diphtheria and tetanus components, the serological assay (method C) can be performed with the same group of animals used for the serological assay of the tetanus vaccine (adsorbed) (2.7.8) when the common immunisation conditions for the diphtheria and the tetanus components (for example, doses, duration) have been demonstrated to be valid for the combined vaccine.
The design of the assays described below uses multiple dilutions for the test and reference preparations. Once the analyst has sufficient experience with this method for a given vaccine, it is possible to apply a simplified model such as a single dilution for both test and reference preparations. Such a model enables the analyst to determine whether the potency of the test preparation is significantly higher than the minimum required, but does not give information on linearity, parallelism and the dose-response curve. The simplified model allows for a considerable reduction in the number of animals required and must be considered by each analyst in accordance with the provisions of the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes.
Where a single-dilution assay is used, production and test consistency over time are monitored via suitable indicators and by carrying out a full multiple-dilution assay periodically, for example every 2 years. For serological assays, suitable indicators to monitor test consistency are:
METHOD A: INTRADERMAL CHALLENGE test in guinea-PIGS
Selection and distribution of the test animals
Use in the test healthy, white guinea-pigs from the same stock and of a size suitable for the prescribed number of challenge sites, the difference in body mass between the heaviest and the lightest animal being not greater than 100 g. Use guinea-pigs of the same sex or with males and females equally distributed between the groups. Distribute the guinea-pigs in not fewer than 6 equal groups; use groups containing a number of animals sufficient to obtain results that fulfil the requirements for a valid assay prescribed below. If the challenge toxin to be used has not been shown to be stable or has not been adequately standardised, include 5 guinea-pigs as unvaccinated controls.
Selection of the challenge toxin
Select a preparation of diphtheria toxin containing 67 to 133 lr/100 in 1 Lf and 25 000 to 50 000 minimal reacting doses for guinea-pig skin in 1 Lf. If the challenge toxin preparation has been shown to be stable, it is not necessary to verify the activity for every assay.
Preparation of the challenge toxin solution
Immediately before use, dilute the challenge toxin with a suitable diluent to obtain a challenge toxin solution containing about 0.0512 Lf in 0.2 mL. Prepare from this a further series of 5 four-fold dilutions containing about 0.0128, 0.0032, 0.0008, 0.0002 and 0.00005 Lf in 0.2 mL.
DILUTION OF THE TEST AND REFERENCE PREPARATIONS
Using a 9 g/L solution of sodium chloride R, prepare dilutions of the vaccine to be examined and of the reference preparation, such that for each, the dilutions form a series differing by not more than 2.5-fold steps and in which the intermediate dilutions, when injected subcutaneously at a dose of 1.0 mL per guinea-pig, will result in an intradermal score of approximately 3 when the animals are challenged.
IMMUNISATION AND CHALLENGE
Allocate the dilutions, 1 to each of the groups of guinea-pigs, and inject subcutaneously 1.0 mL of each dilution into each guinea-pig in the group to which that dilution is allocated. After 28 days, shave both flanks of each guinea-pig and inject 0.2 mL of each of the 6 toxin dilutions intradermally into 6 separate sites on each of the vaccinated guinea-pigs in such a way as to minimise interference between adjacent sites.
Determination of the activity of the challenge toxin
If necessary, inject the unvaccinated control animals with dilutions containing 80, 40, 20, 10 and 5 × 10-6 Lf of the challenge toxin.
Reading and interpretation of results
Examine all injection sites 48 h after injection of the challenge toxin and record the incidence of specific diphtheria erythema. Record also the number of sites free from such reactions as the intra-dermal challenge score. Tabulate the intradermal challenge scores for all the animals receiving the same dilution of vaccine and use those data with a suitable transformation, such as (score)2 or arcsin ((score/6)2), to obtain an estimate of the relative potency for each of the test preparations by parallel-line quantitative analysis.
Requirements for a valid assay
The test is not valid unless:
The test may be repeated but when more than 1 test is performed the results of all valid tests must be combined in the estimate of potency.
METHOD C. DETERMINATION OF ANTIBODIES IN GUINEA-PIGS
Selection and distribution of the test animals
Use in the test healthy guinea-pigs from the same stock, each weighing 250-350 g. Use guinea-pigs of the same sex or with males and females equally distributed between the groups. Distribute the guinea-pigs in not fewer than 6 equal groups; use groups containing a number of animals sufficient to obtain results that fulfil the requirements for a valid assay prescribed below. Use a further group of non-vaccinated guinea-pigs of the same origin to provide a negative serum control. If test consistency has been demonstrated, a reference negative serum control may be used.
Reference preparation
Use a suitable reference preparation such as diphtheria vaccine (adsorbed) BRP or a batch of vaccine shown to be effective in clinical studies, or a batch representative thereof, and which has been calibrated in International Units with reference to diphtheria vaccine (adsorbed) BRP or the International Standard for diphtheria toxoid (adsorbed).
Dilution of the test and reference preparations
Using a 9 g/L solution of sodium chloride R as diluent, prepare serial dilutions of the vaccine to be examined and the reference preparation; series differing by 2.5- to 5-fold steps have been found to be suitable. Use not fewer than 3 dilutions within the range of, for example, 0.5-16 IU/mL for the reference vaccine and within the range of, for example, 1:2 to 1:125 for the vaccine to be examined. Use the dilutions for immunisation preferably within 1 h of preparation. Allocate 1 dilution to each group of guinea-pigs.
Immunisation
Inject subcutaneously to each guinea-pig 1.0 mL of the dilution allocated to its group.
Blood sampling
35-42 days after immunisation, take a blood sample from each vaccinated and control guinea-pig using a suitable method.
Preparation of serum samples
Avoid frequent freezing and thawing of serum samples. To avoid microbial contamination, it is preferable to carry out manipulations in a laminar-flow cabinet.
Determination of antibody titre
Determine the relative antibody titre or score of each serum sample by a suitable immunochemical method (2.7.1). The methods shown below (enzyme-linked immunosorbent assay (ELISA) and Vero cell assay) have been found to be suitable.
Calculation of potency
Calculate the potency of the vaccine to be examined in International Units relative to the reference preparation, using the usual statistical methods (for example, 5.3).
Requirements for a valid assay
The test is not valid unless:
The test may be repeated but when more than 1 test is performed the results of all valid tests must be combined in the estimate of potency.
The following section is published for information.
Assay of diphtheria vaccine (adsorbed): guidelines
method c. DETERMINATION OF ANTIBODIES IN GUINEA-PIGS
Preparation of serum samples
For the preparation of serum samples, the following technique has been found to be suitable. Invert the tubes containing blood samples 6 times and allow to stand at 37 °C for 2 h, then at 4 °C for 2 h. Centrifuge at room temperature at 800 g for 20 min. Transfer the serum to sterile tubes and store at a temperature below -20 °C. At least a 40 per cent yield of serum is obtained by this procedure.
Determination of antibody titre
The ELISA and Vero cell assays shown below are given as examples of immunochemical methods that have been found to be suitable for the determination of antibody titre.
Determination of antibody titre in guinea-pig serum by enzyme-linked immunosorbent assay (ELISA)
Dilutions of test and reference sera are made on ELISA plates coated with diphtheria toxoid. A positive guinea-pig serum control and a negative guinea-pig serum control are included on each plate to monitor the assay performance. Peroxidase-conjugated rabbit or goat antibody directed against guinea-pig-IgG is added, followed by a peroxidase substrate. Optical density is measured and the relative antibody titre is calculated using the usual statistical methods (for example, 5.3).
Reagents and equipment
Method
The description below is given as an example of a suitable plate layout but others may be used. Wells 1A-H are for negative control serum and wells 2A-H and 12A-H are for positive control serum for assay monitoring. Wells 3-11A-H are for test samples.
Coat each well of the ELISA plates with 100 µL of diphtheria toxoid solution (0.5 Lf/mL in carbonate coating buffer pH 9.6). Allow to stand overnight at 4 °C in a humid atmosphere. To avoid temperature gradient effects, do not stack more than 4 plates high. On the following day, wash the plates thoroughly with washing buffer. Block the plates by addition of 100 µL of diluent block buffer to each well. Incubate in a humid atmosphere at 37 °C for 1 h. Wash the plates thoroughly with washing buffer. Place 100 µL of diluent block buffer in each well of the plates, except those of row A. Prepare suitable dilutions of negative control serum, positive control serum (from about 0.01 IU/mL) and test sera. Allocate the negative control serum to column 1, positive control serum to columns 2 and 12 and test sera to columns 3-11 and add 100 µL of each serum to the first 2 wells of the column to which it is allocated. Using a multichannel micropipette, make twofold serial dilutions from row B, down the plate to row H, by transferring 100 µL from one well to the next well. Discard 100 µL from the last row so that all wells contain 100 µL. Incubate at 37 °C for 2 h. Wash thoroughly with washing buffer. Prepare a suitable dilution (a 2000-fold dilution has been found to be suitable) of peroxidase conjugate in diluent block buffer and add 100 µL to each well. Incubate at 37 °C in a humid atmosphere for 1 h. Wash the plates thoroughly with washing buffer. Add 100 µL of peroxidase substrate to each well. Allow to stand at room temperature, protected from light, for 30 min. Read the plates at 405 nm in the same order as addition of substrate was made.
Determination of antibody titre in guinea-pig serum by Vero cell assay
The method used relies either on metabolic inhibition (method 1) or on cytotoxicity (method 2) as the end point, and on either microscopic (cell morphology) or visual (colour) inspection of the cells.
The limit of detection is specific for each antitoxin and is usually between 0.015 IU/mL (method 1) and 0.05 IU/mL (method 2).
The endpoint is taken as the highest serum dilution protecting cells from the diphtheria toxin effect. The antitoxin activity is calculated with respect to guinea-pig or WHO reference standard, and expressed in International Units per millilitre.
Reagents and equipment
Method 1. The diphtheria toxin causes a cytopathogenic effect on Vero cells leading to cellular lysis. Antibodies directed against diphtheria toxin may inhibit this cytopathogenic effect. Consequently, the potency of a diphtheria vaccine may be indirectly determined with the help of this cell culture system if different serum dilutions from immunised animals are cultured with a constant toxin concentration. In the Vero cell assay, yellow colour indicates viable cells, red colour dead cells. When only part of the cells are dead, the colour may be orange.
Reagents and equipment
Vero cells are cultured in tissue culture flasks (for example 75 cm2/250 mL) in an incubator at 36 ± 1 °C, 5 per cent CO2 and 90 per cent relative humidity. Vero cells are first grown in the primary culture medium. After 2-3 days of growth, the primary culture medium is replaced by the maintenance culture medium. When a confluent monolayer is obtained, the culture supernatant is discarded and the cell layer washed gently with modified D-PBS. Add a mixture of 1 volume of trypsin solution and 1 volume of EDTA solution to the flask. Swirl the flask gently and incubate in the CO2 incubator for about 3 min until the cells start to break from the monolayer. Vigorously tap the side of the flask to make the cells fall. Resuspend the cells in 5-6 mL of fresh medium C to obtain a homogeneous suspension. Prepare a cell suspension in medium C containing approximately 1 × 105 cells/mL.
Place 25 µL of medium B in each well except those of column 1. Place 25 µL of the diphtheria guinea-pig antiserum (for vaccines-human use) (positive control serum, working dilution in medium B of 0.40 IU/mL) in wells A1, A2 and A11. Place 25 µL of guinea-pig serum samples in wells B-G of columns 1, 2 and 11. Place 25 µL of negative control serum in row H of columns 1, 2 and 11. Using a multichannel micropipette, make twofold serial dilutions across the plate (from column 2 up to column 10 for rows A-G and up to column 8 for row H). Discard 25 µL from the wells in column 10 in rows A-G, and from well H8.
Reconstitute the diphtheria toxin with saline solution to give a solution of 50 IU/mL. Prepare a 50-fold dilution of this diphtheria toxin dilution in medium B to obtain a working solution of 1.0 IU/mL. Add 25 µL of this working solution to wells A12 and B12 (toxin control). Make twofold serial dilutions by tranferring 25 µL from one well to the next, from well B12 down to H12. Change the tip between each dilution. Discard 25 µL from well H12. Add 25 µL of medium B to wells B12-H12. Then, place 25 µL of the working dilution of the diphtheria toxin (1.0 IU/mL) in each well of rows A-H, from column 1-10, except in wells H9 and H10 (cells only, without serum and without toxin).
Cover the plates with lids or sealer and shake gently. Incubate the plates for at least 2 h in a humid container in a CO2 incubator at 37 °C. Add 200 µL of cell suspension containing 1 x 105 cells/mL to all the wells. Cover the plates with sealer. Incubate at 37 °C for 5 days. Check for microbial contamination by microscopic examination.
Yellow wells are recorded as negative and red wells indicate dead cells and are recorded as positive. A colour between yellow and red indicates a mixture of viable and dead cells and is recorded as positive/negative. The results based on the change in colour can be confirmed by reading viable and dead cells under the microscope.
The potency of the guinea-pig antiserum samples is obtained by comparing the last well of the standard preparation showing complete neutralisation of the toxin, with the last well of the sample demonstrating the same effect. For calculations of potency, it must be remembered that the endpoint may be between a negative well and a positive/negative well.
Method 2: Thiazolyl blue MTT is reduced to a blue/black formazan product by the mitochondrial dehydrogenase of viable cells, and thus serves as a quantitative measure of living cells present, indicating when the toxin has been neutralised by the antitoxin. White or colourless wells indicate absence of viable cells due to insufficient antitoxin to neutralise the toxin.
Reagents and equipment
Vero cells are cultured in tissue culture flasks (for example 75 cm2/250 mL) in an incubator at 36 ± 1 °C, 5 per cent CO2 and 90 per cent relative humidity. Vero cells are grown in the complete culture medium. After 6-7 days of growth, a confluent monolayer is obtained, the culture supernatant is discarded and the cell layer is washed 3 times with trypsin-EDTA: gently pipette out the medium, add 0.5-1 mL of trypsin-EDTA, swirl the flask and tip the trypsin out. Do this twice, and the 3rd time, place the flask in the incubator for 5 min until the cells start to break from the monolayer. Vigorously tap the side of the flask to make the cells fall. Resuspend the cells in 6-25 mL of fresh complete culture medium to obtain a homogeneous suspension. Prepare a cell suspension in complete culture medium containing approximately 4 × 105 cells/mL.
Place 50 µL of complete culture medium in each well except those of column 1. Place 100 µL of diphtheria guinea pig antiserum (for vaccines-human use) (positive control serum, working dilution in complete culture medium of 0.12 IU/mL) in well A1 and 50 µL in well A11. Place 100 µL of guinea pig test serum samples, diluted if necessary, in wells B1-G1. Add 50 µL of the same sample to wells B11-G11 in the corresponding row. Place 100 µL of negative control serum in well H1 and 50 µL in well H11. Using a multi-channel micropipette, make twofold serial dilutions by transferring 50 µL from one well to the next working across the plate (from column 1-10 for rows A-G and from column 1-8 for row H).
Diphtheria toxin of known activity and Lf content is diluted to a suitable working stock containing at least 4 minimum cytopathic doses in complete culture medium. Add 50 µL of the diluted toxin to each well except H9 and H10 (cell control), A11-H11 (serum control) and A12-H12 (toxin control). Add 100 µL of diluted toxin to well A12 and make twofold serial dilutions by transferring 50 µL from one well to the next working down the plate (from well A12-H12). Discard 50 µL from well H12. Add 50 µL of complete medium to wells H9 and H10.
Cover the plates with a lid or sealer and leave for 1 h at room temperature to allow toxin neutralisation to occur. 50 µL of cell suspension containing approximately 4 × 105 cells/mL is added to each well. The plates are sealed and incubated at 37 °C for 6 days. Check for microbial contamination by microscopic examination. 10 µL of thyazolyl blue MTT solution is added to each well. The plates are incubated at 37 °C for a further 2-4 h. Then, the medium is removed and 100 µL of extraction buffer pH 4.7 is added to each well. The plates are incubated at 37 °C and left overnight to aid the extraction process. Once extraction and solubilisation is complete, plates are visually examined or read at 570 nm.
Blue/black wells are recorded as negative (all the cells are alive, toxin neutralisation by antitoxin) and white or colourless wells indicate dead cells (no toxin neutralisation) and are recorded as positive.
The potency of the test antitoxin is obtained by comparing the last well of the reference antitoxin preparation showing neutralisation of the toxin, with the last well of the antitoxin preparation demonstrating the same effect. The neutralising antibody titre of the sample being examined can be calculated by multiplication of the dilution factor with total number of International Units per millilitre of the reference preparation at the end point. The test is valid if all the cells in the toxin control are dead and reference antitoxin gives a neutralisation in at least the first 2 dilutions tested.
2. Assay of pertussis vaccine (Whole Cell)
The potency of pertussis vaccine (whole cell) is determined by comparing the dose necessary to protect mice against the effects of a lethal dose of Bordetella pertussis, administered intracerebrally, with the quantity of a reference preparation, calibrated in International Units, needed to give the same protection.
The International Unit is the activity contained in a stated amount of the International Standard which consists of a quantity of dried pertussis vaccine. The equivalence in International Units of the International Standard is stated by the World Health Organization.
Selection and distribution of the test animals
Use in the test healthy mice less than 5 weeks old of a suitable strain from the same stock, the difference in mass between the heaviest and the lightest being not greater than 5 g. Distribute the mice in 6 groups of not fewer than 16 and 4 groups of 10. The mice must all be of the same sex or the males and females distributed equally between the groups.
Selection of the challenge strain and preparation of the challenge suspension
Select a suitable strain of B. pertussis capable of causing the death of mice within 14 days of intracerebral injection. If more than 20 per cent of the mice die within 48 h of the injection the strain is not suitable. Make one subculture from the strain and suspend the harvested B. pertussis in a solution containing 10 g/L of casein R hydrolysate and 6 g/L of sodium chloride R and having a pH of 7.0 to 7.2 or in another suitable solution. Determine the opacity of the suspension. Prepare a series of dilutions in the same solution and allocate each dilution to a group of 10 mice. Inject intracerebrally into each mouse a dose (0.02 mL or 0.03 mL) of the dilution allocated to its group. After 14 days, count the number of mice surviving in each group. From the results, calculate the expected opacity of a suspension containing 100 LD50 in each challenge dose. For the test of the vaccine to be examined make a fresh subculture from the same strain of B. pertussis and prepare a suspension of the harvested organisms with an opacity corresponding to about 100 LD50 in each challenge dose. Prepare 3 dilutions of the challenge suspension.
Determination of potency
Prepare 3 serial dilutions of the vaccine to be examined and 3 similar dilutions of the reference preparation such that in each the intermediate dilution may be expected to protect about 50 per cent of the mice from the lethal effects of the challenge dose of B. pertussis. Suggested doses are 1/8, 1/40 and 1/200 of the human dose of the vaccine to be examined and 0.5 IU, 0.1 IU and 0.02 IU of the reference preparation, each dose being contained in a volume not exceeding 0.5 mL. Allocate the 6 dilutions, one to each of the groups of not fewer than 16 mice, and inject intraperitoneally into each mouse one dose of the dilution allocated to its group. After 14 - 17 days inject intracerebrally into each animal in the groups of not fewer than 16, one dose of the challenge suspension. Allocate the challenge suspension and the 3 dilutions made from it, one to each of the groups of 10 mice, and inject intracerebrally one dose of each suspension into each mouse in the group to which that suspension is allocated. Exclude from consideration any mice that die within 48 h of challenge. Count the number of mice surviving in each of the groups after 14 days. Calculate the potency of the vaccine to be examined relative to the potency of the reference preparation on the basis of the numbers of animals surviving in each of the groups of not fewer than 16.
The test is not valid unless:
The test may be repeated but when more than one test is performed the results of all valid tests must be combined.
3. Assay of tetanus vaccine (adsorbed)
The potency of tetanus vaccine is determined by administration of the vaccine to animals (guinea-pigs or mice) followed either by challenge with tetanus toxin (method A or B) or by determination of the titre of antibodies against tetanus toxoid in the serum of the guinea-pigs (method C). In both cases, the potency of the vaccine is calculated by comparison with a reference vaccine, calibrated in International Units. For methods A and B, in countries where the paralysis method is not obligatory, the LD50 method may be used. For the LD50 method, the number of animals and the procedure are identical to those described for the paralysis method, but the end-point is the death of the animal rather than paralysis.
The International Unit is the activity contained in a stated amount of the International Standard for tetanus toxoid (adsorbed). The equivalence in International Units of the International Standard is stated by the World Health Organization.
Tetanus vaccine (adsorbed) BRP is calibrated in International Units with reference to the International Standard.
The method chosen for the assay of tetanus vaccine (adsorbed) depends on the intended purpose. Method A or B is used:
Method A or B may also be used for the routine assay of batches of vaccine, but in the interests of animal welfare, method C is used wherever possible.
Method C may be used, except as specified under 1 and 2 above, after verification of the suitability of the method for the product. For this purpose, a suitable number of batches (usually 3) are assayed by method C and method A or B. Where different vaccines (monovalent or combinations) are prepared from tetanus toxoid of the same origin and with comparable levels (expressed in Lf/mL) of the same tetanus toxoid, suitability demonstrated for the combination with the highest number of components can be assumed to be valid for combinations with fewer components and for monovalent vaccines. Any combinations containing a whole-cell pertussis component or containing haemophilus type b conjugate vaccine with tetanus toxoid in the same vial must always be assessed separately.
For combinations containing diphtheria and tetanus components, the serological assay (method C) can be performed with the same group of animals used for the serological assay of the diphtheria vaccine (adsorbed) (2.7.6) when the common immunisation conditions for the tetanus and the diphtheria components (for example, doses, duration) have been demonstrated to be valid for the combined vaccine.
The design of the assays described below uses multiple dilutions for the test and reference preparations. Based on the potency data obtained in multiple-dilution assays, it may be possible to reduce the number of animals needed to obtain a statistically significant result by applying a simplified model such as a single dilution for both test and reference preparations. Such a model enables the analyst to determine whether the potency of the test preparation is significantly higher than the minimum required, but does not give information on the dose-response curves and their linearity, parallelism and significant slope. The simplified model allows for a considerable reduction in the number of animals required and must be considered by each analyst in accordance with the provisions of the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes.
Where a single-dilution assay is used, production and test consistency over time are monitored via suitable indicators and by carrying out a full multiple-dilution assay periodically, for example every 2 years. For serological assays, suitable indicators to monitor test consistency are:
METHOD A. CHALLENGE TEST IN GUINEA-PIGS
Selection and distribution of the test animals
Use in the test healthy guinea-pigs from the same stock, each weighing 250-350 g. Use guinea-pigs of the same sex or with males and females equally distributed between the groups. Distribute the guinea-pigs in not fewer than 6 equal groups; use groups containing a number of animals sufficient to obtain results that fulfil the requirements for a valid assay prescribed below. If the activity of the challenge toxin has to be determined, include 3 further groups of 5 guinea-pigs as unvaccinated controls.
Selection of the challenge toxin
Select a preparation of tetanus toxin containing not less than 50 times the 50 per cent paralytic dose per millilitre. If the challenge toxin preparation has been shown to be stable, it is not necessary to verify the paralytic dose for every assay.
Preparation of the challenge toxin solution
Immediately before use, dilute the challenge toxin with a suitable diluent (for example, peptone buffered saline solution pH 7.4) to obtain a stable challenge toxin solution containing approximately 50 times the 50 per cent paralytic dose per millilitre. If necessary, use portions of the challenge toxin solution diluted 1 to 16, 1 to 50 and 1 to 160 with the same diluent to determine the activity of the toxin.
Dilution of the test and reference preparations
Using a 9 g/L solution of sodium chloride R, prepare dilutions of the vaccine to be examined and of the reference preparation, such that for each, the dilutions form a series differing by not more than 2.5-fold steps and in which the intermediate dilutions, when injected subcutaneously at a dose of 1.0 mL per guinea-pig, protect approximately 50 per cent of the animals from the paralytic effects of the subcutaneous injection of the quantity of tetanus toxin prescribed for this test.
Immunisation and challenge
Allocate the dilutions, 1 to each of the groups of guinea-pigs, and inject subcutaneously 1.0 mL of each dilution into each guinea-pig in the group to which that dilution is allocated. After 28 days, inject subcutaneously into each animal 1.0 mL of the challenge toxin solution (containing 50 times the 50 per cent paralytic dose).
Determination of the activity of the challenge toxin
If necessary, allocate the 3 dilutions made from the challenge toxin solution, 1 to each of the 3 groups of 5 guinea-pigs, and inject subcutaneously 1.0 mL of each solution into each guinea-pig in the group to which that solution is allocated. The activity and stability of the challenge toxin are determined by carrying out a suitable number of determinations of the 50 per cent paralytic dose. It is then not necessary to repeat the determination for each assay.
Reading and interpretation of results
Examine the guinea-pigs twice daily. Remove and euthanise all animals showing definite signs of tetanus paralysis. Count the number of guinea-pigs without paralysis 5 days after injection of the challenge toxin. Calculate the potency of the vaccine to be examined relative to the potency of the reference preparation on the basis of the proportion of challenged animals without paralysis in each group of vaccinated guinea-pigs, using the usual statistical methods (for example, 5.3).
Requirements for a valid assay
The test is not valid unless:
The test may be repeated but when more than 1 test is performed the results of all valid tests must be combined in the estimate of potency.
METHOD B. CHALLENGE TEST IN MICE
Selection and distribution of the test animals
Use in the test healthy mice from the same stock, about 5 weeks old and from a strain shown to be suitable. Use mice of the same sex or with males and females equally distributed between the groups. Distribute the mice in not fewer than 6 equal groups; use groups containing a number of animals sufficient to obtain results that fulfil the requirements for a valid assay prescribed below. If the challenge toxin to be used has not been shown to be stable or has not been adequately standardised, include 3 further groups of not fewer than 5 mice to serve as unvaccinated controls.
Selection of the challenge toxin
Select a preparation of tetanus toxin containing not less than 100 times the 50 per cent paralytic dose per millilitre. If the challenge toxin preparation has been shown to be stable, it is not necessary to verify the paralytic dose for every assay.
Preparation of the challenge toxin solution
Immediately before use, dilute the challenge toxin with a suitable diluent (for example, peptone buffered saline solution pH 7.4) to obtain a stable challenge toxin solution containing approximately 50 times the 50 per cent paralytic dose in 0.5 mL. If necessary, use portions of the challenge toxin solution diluted 1 to 16, 1 to 50 and 1 to 160 with the same diluent to determine the activity of the toxin.
Dilution of the test and reference preparations
Using a 9 g/L solution of sodium chloride R, prepare dilutions of the vaccine to be examined and of the reference preparation, such that for each, the dilutions form a series differing by not more than 2.5-fold steps and in which the intermediate dilutions, when injected subcutaneously at a dose of 0.5 mL per mouse, protect approximately 50 per cent of the animals from the paralytic effects of the subcutaneous injection of the quantity of tetanus toxin prescribed for this test.
Immunisation and challenge
Allocate the dilutions, 1 to each of the groups of mice, and inject subcutaneously 0.5 mL of each dilution into each mouse in the group to which that dilution is allocated. After 28 days, inject subcutaneously into each animal 0.5 mL of the challenge toxin solution (containing 50 times the 50 per cent paralytic dose).
Determination of the activity of the challenge toxin
If necessary, allocate the 3 dilutions made from the challenge toxin solution, 1 to each of the 3 groups of not fewer than 5 mice, and inject subcutaneously 0.5 mL of each solution into each mouse in the group to which that solution is allocated.
Reading and interpretation of results
Examine the mice twice daily. Remove and euthanise all animals showing definite signs of tetanus paralysis. Count the number of mice without paralysis 4 days after injection of the challenge toxin. Calculate the potency of the vaccine to be examined relative to the potency of the reference preparation on the basis of the proportion of challenged animals without paralysis in each group of vaccinated mice, using the usual statistical methods (for example, 5.3).
Requirements for a valid assay
The test is not valid unless:
The test may be repeated but when more than 1 test is performed the results of all valid tests must be combined in the estimate of potency.
METHOD C. DETERMINATION OF ANTIBODIES IN GUINEA-PIGS
Selection and distribution of the test animals
Use in the test healthy guinea-pigs from the same stock, each weighing 250-350 g. Use guinea-pigs of the same sex or with males and females equally distributed between the groups. Distribute the guinea-pigs in not fewer than 6 equal groups; use groups containing a number of animals sufficient to obtain results that fulfil the requirements for a valid assay prescribed below. Use a further group of non-vaccinated guinea-pigs of the same origin to provide a negative serum control. If test consistency has been demonstrated, a reference negative serum control may be used.
Reference preparation
Use a suitable reference preparation such as tetanus vaccine (adsorbed) BRP or a batch of vaccine shown to be effective in clinical studies, or a batch representative thereof, and which has been calibrated in International Units with reference to tetanus vaccine (adsorbed) BRP or the International Standard for tetanus toxoid (adsorbed).
Dilution of the test and reference preparations
Using a 9 g/L solution of sodium chloride R as diluent, prepare serial dilutions of the vaccine to be examined and the reference preparation; series differing by 2.5- to 5-fold steps have been found to be suitable. Use not fewer than 3 dilutions within the range of, for example, 0.5-16 IU/mL for each series. Use the dilutions for immunisation preferably within 1 h of preparation. Allocate 1 dilution to each group of guinea-pigs.
Immunisation
Inject subcutaneously to each guinea-pig 1.0 mL of the dilution allocated to its group.
Blood sampling
35-42 days after immunisation, take a blood sample from each vaccinated and control guinea-pig using a suitable method.
Preparation of serum samples
Avoid frequent freezing and thawing of serum samples. To avoid microbial contamination, it is preferable to carry out manipulations in a laminar-flow cabinet.
Determination of antibody titre
Determine the relative antibody titre or score of each serum sample by a suitable immunochemical method (2.7.1). The methods shown below (enzyme-linked immunosorbent assay (ELISA) and toxin-binding inhibition (ToBI)) have been found to be suitable.
Calculation of potency
Calculate the potency of the vaccine to be examined in International Units relative to the reference preparation, using the usual statistical methods (for example, 5.3).
Requirements for a valid assay
The test is not valid unless:
The test may be repeated but when more than 1 test is performed the results of all valid tests must be combined in the estimate of potency.
The following section is published for information.
Assay of tetanus vaccine (adsorbed): guidelines
METHOD A. CHALLENGE TEST IN GUINEA-PIGS
Reading and interpretation of results
In order to minimise suffering in the test animals, it is recommended to note the degree of paralysis on a scale such as that shown below. The scale gives typical signs when subcutaneous injection of the challenge toxin is made mid-ventrally, directly behind the sternum with the needle pointing towards the neck of the guinea-pig. Grade T3 is taken as the end-point, but with experience grade T2 can be used instead. Tetanus toxin produces in at least 1 of the forelimbs paralysis that can be recognised at an early stage. The tetanus grades in guinea-pigs are characterised by the following signs:
METHOD B. CHALLENGE TEST IN MICE
Reading and interpretation of results
In order to minimise suffering in the test animals, it is recommended to note the degree of paralysis on a scale such as that shown below. The scale gives typical signs when injection of the challenge toxin is made in the dorsal region, close to one of the hind legs. Grade T3 is taken as the end-point, but with experience grade T2 can be used instead. Tetanus toxin produces in the toxin-injected hind leg paresis followed by paralysis that can be recognised at an early stage. The tetanus grades in mice are characterised by the following signs:
METHOD C. DETERMINATION OF ANTIBODIES IN GUINEA-PIGS
Preparation of serum samples
For the preparation of serum samples, the following technique has been found to be suitable. Invert the tubes containing blood samples 6 times and allow to stand at 37 °C for 2 h, then at 4 °C for 2 h. Centrifuge at room temperature at 800 g for 20 min. Transfer the serum to sterile tubes and store at a temperature below -20 °C. At least a 40 per cent yield of serum is obtained by this procedure.
Determination of antibody titre
The ELISA and ToBI tests shown below are given as examples of immunochemical methods that have been found to be suitable for the determination of antibody titre.
Determination of antibody titre in guinea-pig serum by enzyme-linked immunosorbent assay (ELISA)
Dilutions of test and reference sera are made on ELISA plates coated with tetanus toxoid. A positive guinea-pig serum control and a negative guinea-pig serum control are included on each plate to monitor the assay performance. Peroxidase-conjugated rabbit or goat antibody directed against guinea-pig-IgG is added, followed by a peroxidase substrate. Optical density is measured and the relative antibody titre is calculated using the usual statistical methods (for example, 5.3).
Reagents and equipment
Method
The description below is given as an example of a suitable plate layout but others may be used. Wells 1A-H are for negative control serum and wells 2A-H and 12A-H are for positive control serum for assay monitoring. Wells 3-11A-H are for test samples.
Coat each well of the ELISA plates with 100 µL of tetanus toxoid solution (0.5 Lf/mL in carbonate coating buffer pH 9.6). Allow to stand overnight at 4 °C in a humid atmosphere. To avoid temperature gradient effects, do not stack more than 4 plates high. On the following day, wash the plates thoroughly with washing buffer. Block the plates by addition of 100 µL of diluent block buffer to each well. Incubate in a humid atmosphere at 37 °C for 1 h. Wash the plates thoroughly with washing buffer. Place 100 µL of diluent block buffer in each well of the plates, except those of row A. Prepare suitable dilutions of negative control serum, positive control serum (from about 0.01 IU/mL) and test sera. Allocate the negative control serum to column 1, positive control serum to columns 2 and 12 and test sera to columns 3-11 and add 100 µL of each serum to the first 2 wells of the column to which it is allocated. Using a multichannel micropipette, make twofold serial dilutions from row B down the plate to row H, by transferring 100 µL from one well to the next. Discard 100 µL from the last row so that all wells contain 100 µL. Incubate at 37 °C for 2 h. Wash thoroughly with washing buffer. Prepare a suitable dilution (a 2000-fold dilution has been found to be suitable) of peroxidase conjugate in diluent block buffer and add 100 µL to each well. Incubate at 37 °C in a humid atmosphere for 1 h. Wash the plates thoroughly with washing buffer. Add 100 µL of peroxidase substrate to each well. Allow to stand at room temperature, protected from light, for 30 min. Read the plates at 405 nm in the same order as addition of substrate was made.
Determination of antibody titre in guinea-pig serum by toxin- or toxoid-binding inhibition (ToBI)
Tetanus toxin or toxoid is added to serial dilutions of test and reference sera; the serum/antigen mixtures are incubated overnight. To determine unbound toxin or toxoid, the mixtures are transferred to an ELISA plate coated with tetanus antitoxin. Peroxidase-conjugated equine anti-tetanus IgG is added followed by a peroxidase substrate. Optical density is measured and the antibody titre is calculated using the usual statistical methods (for example, 5.3). A positive control serum and a negative control serum are included on each plate to monitor assay performance.
Reagents and equipment
Method
Block the microtitre plates by placing in each well 150 µL of block buffer. Cover the plates with a lid or sealer. Incubate in a humid atmosphere at 37 °C for 1 h. Wash the plates thoroughly with washing solution. Place 100 µL of PBS in each well. Place 100 µL of reference guinea-pig tetanus antitoxin in the first well of a row. Place 100 µL of undiluted test sera in the first well of the required number of rows. Using a multichannel micropipette, make twofold serial dilutions across the plate (up to column 10), by transferring 100 µL from one well to the next. Discard 100 µL from the last column so that all wells contain 100 µL. Prepare a 0.1 Lf/mL solution of tetanus toxin or toxoid using PBS as diluent. Add 40 µL of this solution to each well except those of column 12. The wells of column 11 are a positive control. Add 40 µL of PBS to the wells of column 12 (negative control). Shake the plates gently and cover them with lids. Coat the ELISA plates: immediately before use make a suitable dilution of equine anti-tetanus IgG in carbonate buffer pH 9.6 and add 100 µL to each well. Incubate the 2 series of plates overnight in a humid atmosphere at 37 °C. To avoid temperature gradient effects, do not stack more than 4 plates high. Cover the plates with lids. On the following day, wash the ELISA plates thoroughly with washing solution. Block the plates by placing in each well 125 µL of block buffer. Incubate at 37 °C in a humid atmosphere for 1 h. Wash the plates thoroughly with washing solution. Transfer 100 µL of the pre-incubation mixture from the polystyrene plates to the corresponding wells of the ELISA plates, starting with column 12 and then continuing from 1 to 11. Cover the plates with a lid. Incubate at 37 °C in a humid atmosphere for 2 h. Wash the ELISA plates thoroughly with washing solution. Make a suitable dilution (a 4000-fold dilution has been found to be suitable) of the peroxidase-conjugated equine anti-tetanus IgG in diluent buffer. Add 100 µL of the dilution to each well and cover the plates with a lid. Incubate at 37 °C in a humid atmosphere for 1.5 h. Wash the ELISA plates thoroughly with washing solution. Add 100 µL of peroxidase substrate to each well. A blue colour develops. Incubate the plates at room temperature. Stop the reaction at a given time (within 10 min) by the addition of 100 µL of 2 M sulfuric acid to each well in the same order as the addition of substrate. The colour changes from blue to yellow. Measure the absorbance at 450 nm immediately after addition of the sulfuric acid or maintain the plates in the dark until reading.
4. Assay of hepatitis A vaccine
The assay of hepatitis A vaccine is carried out either in vitro, by an immunochemical determination of antigen content (method A), or in vivo, by comparing in given conditions its capacity to induce specific antibodies in mice with the same capacity of a reference preparation (method B).
METHOD A. IN VITRO ASSAY
The hepatitis A antigen content is determined by a suitable immunochemical method (2.7.1) such as enzyme-linked immunosorbent assay (ELISA).
Hepatitis A vaccine (inactivated, non-adsorbed) BRP may be used as a reference preparation.
The following method is given as an example of an immunochemical method that has been found to be suitable. Perform an ELISA assay for the determination of hepatitis A antigen content using monoclonal antibodies specific for the detection of a hepatitis A epitope that induces neutralising antibodies.
Materials
ELISA microtitre plates 96 wells.
Hepatitis A virus coating reagent for ELISA BRR .
Hepatitis A vaccine ELISA detection antibodies set BRR , composed of anti-hepatitis A virus primary detection antibody BRR and conjugated secondary detection antibody BRR .
Hepatitis A vaccine (inactivated, non-adsorbed) BRP.
Carbonate-bicarbonate coating buffer (0.05 M) pH 9.6 Solution 1: dissolve 2.1 g of sodium hydrogen carbonate R in 500 mL of highly purified water R. Solution 2: dissolve 7.16 g of sodium carbonate R in 500 mL of highly purified water R. Mix 70 mL of solution 1 and about 30 mL of solution 2, adjusting the pH with solution 2 if necessary. Filter through a membrane filter (nominal pore size 0.22 µm) and prepare appropriate aliquots in sterile tubes. Store at 2-8 °C for up to 5 months.
Blocking buffer Solution 1: dissolve 4.2 g of sodium hydrogen carbonate R in 1000 mL of highly purified water R. Solution 2: dissolve 5.3 g of sodium carbonate R in 1000 mL of highly purified water R. Mix 630 mL of solution 1 and 270 mL of solution 2, add 50 g of sucrose R and stir until dissolution is complete. Add 3 g of bovine albumin R (pour small quantities to avoid lumps) and stir until dissolution is complete, avoiding foaming. Adjust to pH 9.6 carefully with solution 2. Prepare appropriate aliquots in sterile tubes. Store at -20 °C for up to 5 months.
PBS buffer pH 7.5 ± 0.3 Dissolve 0.2 g of potassium dihydrogen phosphate R, 0.2 g of potassium chloride R, 8.0 g of sodium chloride R and 1.15 g of disodium hydrogen phosphate dodecahydrate R in 1000 mL of highly purified water R. Store at 2-8 °C for up to 1 month.
Polysorbate 20 solution Dilute 5 mL of polysorbate 20 R to 100 mL with highly purified water R. Store at 2-8 °C for up to 1 month.
PBS-T buffer Immediately before use, add 1 mL of the polysorbate 20 solution to 1 L of the PBS buffer pH 7.5 ± 0.3.
PBS-B-T buffer Dissolve 2.5 g of bovine albumin R in 400 mL of the PBS-T buffer (pour small quantities of bovine albumin to avoid lumps) and stir until dissolution is complete, avoiding foaming. Adjust to pH 7.25 with 4 M sodium hydroxide R and dilute to 500 mL with the PBS-T buffer. Prepare appropriate aliquots in sterile tubes. Store at -20 °C for up to 5 months.
Chromogenic substrate Immediately before use, dissolve at room temperature diammonium 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonate) R (ABTS) according to the manufacturer′s instructions.
Stop solution Dissolve 1 g of sodium dodecyl sulfate R in highly purified water R and dilute to 100 mLwith the same solvent. Store at room temperature for up to 1 month, protected from light.
Method
Dilute an appropriate volume of hepatitis A virus coating reagent for ELISA BRR in an appropriate volume of the carbonate-bicarbonate coating buffer (0.05 M) pH 9.6. Transfer 100 µL of the solution to each well of the microtitre plates. Seal the plates and incubate for 3 h ± 5 min at 37 ± 2 °C in a humidified atmosphere. Store at 5 ± 3 °C overnight in a humidified atmosphere. On the following day, empty the plates by inverting them on absorbent paper and add 200 µL of the blocking buffer to each well. Incubate for 45 ± 5 min at 37 ± 2 °C in a humidified atmosphere. Empty the plates by inverting them on absorbent paper. Seal the plates and store at-20 °C for up to 1 month.
For adsorbed vaccines, a pre-treatment or desorption step is necessary before dilution. The treatment step is considered as a pre-dilution. Prepare 2 independent dilution series in the PBS-B-T buffer for each test sample and for the reference preparation. A 2-fold serial dilution has been found suitable. Thaw an appropriate number of coated plates for the assay. Wash the plates 3 times with the PBS-T buffer. Tap the plates gently on absorbent paper to drain off the washing buffer. For the test samples and the reference preparation, transfer 100 µL of each sample (pre-dilution and serial dilutions) to separate wells of the plate following the appropriate plate layout. For blanks, transfer 100 µL of the PBS-B-T buffer per well. Seal the plates and incubate at 37 ± 2 °C in a humidified atmosphere for 1.5 h ± 5 min. Discard the incubation mix and wash the plates 3 times with the PBS-T buffer. Tap the plates gently on absorbent paper to drain off the washing buffer.
Immediately before use, dilute an appropriate volume of anti-hepatitis A virus primary detection antibody BRR in an appropriate volume of the PBS-B-T buffer. Transfer 100 µL of the solution to each well. Seal the plates and incubate at 37 ± 2 °C in a humidified atmosphere for 1 h ± 3 min. Discard the incubation mix and wash the plates 3 times with the PBS-T buffer. Tap the plates gently on absorbent paper to drain off the washing buffer.
Immediately before use, dilute an appropriate volume of conjugated secondary detection antibody BRR in an appropriate volume of the PBS-B-T buffer. Transfer 100 µL of the solution to each well. Seal the plates and incubate at 37 ± 2 °C in a humidified atmosphere for 1 h ± 3 min. Discard the incubation mix and wash the plates 3 times with the PBS-T buffer. Tap the plates gently on absorbent paper to drain off the washing buffer.
Add 100 µL of the chromogenic substrate to each well. Seal the plates and incubate at 37 ± 2 °C in a humidified atmosphere for 30 ± 2 min. Stop the reaction by adding 50 µL of the stop solution to each well. Read the absorbance immediately at 405 nm.
Calculation
Calculate the potency of the vaccine to be examined relative to hepatitis A vaccine (inactivated, non-adsorbed) BRP, using the usual statistical methods (5.3).
Validity conditions
The test is not valid unless:
Method B. IN VIVO ASSAY
The test in mice shown below is given as an example of a method that has been found suitable for a given vaccine; other validated methods may also be used.
Selection and distribution of the test animals
Use in the test healthy mice from the same stock, about 5 weeks old and from a strain shown to be suitable. Use animals of the same sex. Distribute the animals in at least 7 equal groups of a number suitable for the requirements of the assay.
Determination of potency of the vaccine to be examined
Using a 9 g/L solution of sodium chloride R containing the aluminium adjuvant used for the vaccine, prepare at least 3 dilutions of the vaccine to be examined and matching dilutions of the reference preparation. Allocate the dilutions one to each of the groups of animals and inject subcutaneously not more than 1.0 mL of each dilution into each animal in the group to which that dilution is allocated. Maintain a group of unvaccinated controls, injected subcutaneously with the same volume of diluent. After 28-32 days, anaesthetise and bleed all animals, keeping the individual sera separate. Assay the individual sera for specific antibodies against hepatitis A virus by a suitable immunochemical method (2.7.1).
Calculations
Carry out the calculations by the usual statistical methods for an assay with a quantal response (5.3).
From the distribution of reaction levels measured on all the sera in the unvaccinated group, determine the maximum reaction level that can be expected to occur in an unvaccinated animal for that particular assay. Any response in vaccinated animals that exceeds this level is by definition a seroconversion.
Make a suitable transformation of the percentage of animals showing seroconversion in each group (for example, a probit transformation) and analyse the data according to a parallel-line log dose-response model. Determine the potency of the test preparation relative to the reference preparation.
Validity conditions
The test is not valid unless:
Potency requirement
The upper confidence limit (P = 0.95) of the estimated relative potency is not less than 1.0.
5. Assay of hepatitis B vaccine (rDNA)
The assay of hepatitis B vaccine (rDNA) is carried out either in vivo, by comparing in given conditions its capacity to induce specific antibodies against hepatitis B surface antigen (HBsAg) in mice or guinea-pigs with the same capacity of a reference preparation, or in vitro, by an immunochemical determination of the antigen content.
IN VIVO ASSAY
Selection and distribution of the test animals
Use in the test healthy mice from the same stock, about 5 weeks old. The strain of mice used for this test must give a significant slope for the dose-response curve to the antigen; mice with haplotype H-2q or H-2d are suitable. Healthy guinea-pigs weighing 300 g to 350 g (about 7 weeks old) from the same stock are also suitable. Use animals of the same sex. Distribute the animals in at least 7 equal groups of a number appropriate to the requirements of the assay.
Determination of potency of the vaccine to be examined
Using a 9 g/L solution of sodium chloride R containing the aluminium adjuvant used for the vaccine or another appropriate diluent, prepare at least 3 dilutions of the vaccine to be examined and matching dilutions of the reference preparation. Allocate the dilutions, 1 to each of the groups of animals, and inject intraperitoneally not more than 1.0 mL of each dilution into each animal in the group to which that dilution is allocated. One group of animals remains unvaccinated and is injected intraperitoneally with the same volume of diluent. After an appropriate time interval (for example, 4-6 weeks), anaesthetise and bleed the animals, keeping the individual sera separate. Assay the individual sera for specific antibodies against HBsAg by a suitable immunochemical method (2.7.1).
Calculations
Calculations are carried out by the usual statistical methods for an assay with a quantal response (5.3).
From the distribution of reaction levels measured on all the sera in the unvaccinated group, the maximum reaction level that can be expected to occur in an unvaccinated animal for that particular assay is determined. Any response in vaccinated animals that exceeds this level is by definition a seroconversion.
Make a suitable transformation of the percentage of animals showing seroconversion in each group (for example, a probit transformation) and analyse the data according to a parallel-line log dose-response model. Determine the potency of the test preparation relative to the reference preparation.
Validity conditions
The test is not valid unless:
Potency requirement
The upper confidence limit (P = 0.95) of the estimated relative potency is not less than 1.0.
IN VITRO ASSAY
Carry out an immunochemical determination (2.7.1) of antigen content with acceptance criteria validated against the in vivo test.
Enzyme-linked immunosorbent assay (ELISA) and radio-immunoassay (RIA) using monoclonal antibodies specific for protection-inducing epitopes of HBsAg have been shown to be suitable. Suitable numbers of dilutions of the vaccine to be examined and the reference preparation are used and a parallel-line model is used to analyse the data, which may be suitably transformed. Kits for measuring HBsAg in vitro are commercially available and it is possible to adapt their test procedures for use as an in vitro potency assay.
The acceptance criteria are approved for a given reference preparation by the competent authority in light of the validation data.
6. Assay of pertussis vaccine (acellular)
The capacity of the vaccine to induce the formation of specific antibodies in mice or guinea-pigs is compared with the same capacity of a reference preparation examined in parallel; antibodies are determined using a suitable immunochemical method (2.7.1) such as enzyme-linked immunosorbent assay (ELISA).
For combinations containing pertussis components together with diphtheria and tetanus components, the serological assay in guinea-pigs can be performed with the same group of animals used for the serological assay of diphtheria vaccine (adsorbed) (2.7.6) and of tetanus vaccine (adsorbed) (2.7.8) when the common immunisation conditions for all components (for example, doses, duration) have been demonstrated to be valid for the combined vaccine. The guinea-pig model allows for a further reduction in the number of animals required and must be considered by each analyst in accordance with the provisions of the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes.
The design of the assays A and B described below uses multiple dilutions for the test and reference preparations. After validation for a given vaccine, it is possible to apply a simplified model such as a single dilution for both test and reference preparations. Such a model enables the analyst to determine whether the immunogenicity of the test preparation is comparable to the reference vaccine, but does not give information on linearity or parallelism of the dose-response curves.
For serological assays, suitable indicators to monitor test consistency are:
Where a single-dilution assay is used, production and test consistency over time are monitored via suitable indicators and by carrying out a full multiple-dilution assay periodically, for example every 2 years.
METHOD A. SEROLOGY IN MICE
Reference vaccine
A batch of vaccine shown to be effective in clinical trials or a batch representative thereof is used as a reference vaccine. For the preparation of a representative batch, strict adherence to the production process used for the batch tested in clinical trials is necessary. The stability of the reference vaccine shall be monitored and documented.
Reference antiserum
A reference antiserum of assigned activity is used in the test and serves as the basis for calculation of the antibody levels in the test sera. Bordetella pertussis mouse antiserum BRP is suitable for use as a reference antiserum.
The following test model is given as an example of a method that has been found to be satisfactory.
Selection and distribution of the test animals
Use in the test healthy mice (for example, CD1 strain) of the same stock, about 5 weeks old. Distribute the animals in 6 groups of a number appropriate to the requirements of the assay. Use 3 dilutions of the vaccine to be examined and 3 dilutions of a reference preparation and attribute each dilution to a group of mice. Inject intraperitoneally or subcutaneously into each mouse 0.5 mL of the dilution attributed to its group. During validation studies a further group of mice may be used as a negative control by injecting the animals with diluent alone.
Collection of serum samples
4-5 weeks after vaccination, bleed the mice individually under anaesthesia. Store the sera at -20 °C until used for antibody determination.
Antibody determination
Assay the individual sera for content of specific antibodies to each acellular pertussis antigen using a validated method such as the ELISA test shown below.
ELISA test
Microtitre plates (poly(vinyl chloride) or polystyrene as appropriate for the specific antigen) are coated with the purified antigen at a concentration of 100 ng per well. After washing, unreacted sites are blocked by incubating the plates with a solution of bovine serum albumin and then washed. 2-fold dilutions of sera from individual mice immunised with test or reference vaccines are made on the plates. Reference antiserum is included on each plate. After incubation at 22-25 °C for 1 h, the plates are washed. A suitable solution of enzyme-conjugated anti-mouse IgG antibody is added to each well and incubated at 22-25 °C for 1 h. After washing, a chromogenic substrate is added from which the bound enzyme conjugate liberates a chromophore that can be quantified by measurement of absorbance (2.2.25).
Calculations
The antibody titres in the sera of mice immunised with reference and test vaccines are calculated for each acellular pertussis antigen using the reference antiserum, and from the values obtained the relative potency of the test vaccine in relation to the reference vaccine is calculated by the usual statistical methods (5.3).
The assay is not valid unless:
METHOD B. SEROLOGY IN GUINEA-PIGS
Selection and distribution of the test animals
Use in the test healthy guinea-pigs from the same stock, each weighing 250-350 g. Use guinea-pigs of the same sex or with males and females equally distributed between the groups. Distribute the guinea-pigs in not fewer than 6 equal groups; use groups containing a number of animals sufficient to obtain results that fulfil the requirements for a valid assay prescribed below. During validation studies a further group of guinea-pigs is used as a negative control by injecting the animals with diluent alone.
Reference vaccine
A batch of vaccine shown to be effective in clinical trials or a batch representative thereof is used as a reference vaccine. For the preparation of a representative batch, strict adherence to the production process used for the batch tested in clinical trials is necessary. The stability of the reference vaccine shall be monitored and documented.
Reference antiserum
An in-house guinea-pig reference antiserum of assigned activity is used in the test and serves as the basis for calculation of the antibody levels in the test sera.
Dilution of the test and reference preparations
Using a 9 g/L solution of sodium chloride R as diluent, prepare serial dilutions of the vaccine to be examined and the reference preparation; series differing by 2.5- to 5-fold steps have been found to be suitable. Use not fewer than 3 dilutions within the range found to be suitable for all the components in the vaccine to be examined. Use the dilutions for immunisation preferably within 1 h of preparation. Allocate 1 dilution to each group of guinea-pigs.
Immunisation
Inject subcutaneously into each guinea-pig 1.0 mL of the dilution allocated to its group.
Blood sampling
35-42 days (5-6 weeks) after immunisation, take a blood sample from each vaccinated and negative control guinea-pig using a suitable method. Store the sera at -20 °C until used for antibody determination. Avoid frequent freezing and thawing of serum samples.
Antibody determination
Assay the individual sera for content of specific antibodies to each acellular pertussis antigen using a validated method such as the ELISA test shown below.
ELISA test
Suitable 96-well microtitre plates are coated with the purified antigens (e.g. pertussis toxin (PT), pertactin (PRN), filamentous haemagglutinin (FHA) and/or fimbrial agglutinogens (Fim 2/3)) representing components in the combined vaccine at a concentration of 200-400 ng per well. After washing, unreacted sites are blocked by incubating the plates with a suitable blocking buffer and then washed. 2-fold dilutions of sera from individual guinea-pigs immunised with test or reference vaccines are made on the plates. Reference antiserum is included on each plate. After incubation at 37 °C for 1 h, the plates are washed. A suitable solution of enzyme-conjugated anti-guinea-pig IgG antibody is added to each well and incubated at 37 °C for 1 h. After washing, a chromogenic substrate is added from which the bound enzyme conjugate liberates a chromophore that can be quantified by measurement of absorbance (2.2.25).
Calculations
The antibody titres in the sera of guinea-pigs immunised with reference and test vaccines are calculated for each acellular pertussis antigen using the reference antiserum, and from the values obtained the relative potency of the test vaccine in relation to the reference vaccine is calculated by the usual statistical methods (5.3).
The assay is not valid unless:
Assay of pertussis vaccine (acellular): guidelines
METHOD B. DETERMINATION OF ANTIBODIES IN GUINEA-PIGS
The ELISA shown below is given as an example of an immunochemical method that has been found to be suitable.
Determination of antibody titre by ELISA method for pertussis toxin (PT), filamentous haemagglutinin (FHA), fimbrial agglutinogens (Fim 2/3) and pertactin (PRN)
2-fold dilutions of sera from test and reference vaccines are made on ELISA plates coated with acellular pertussis antigens (PRN, PT, FHA or Fim 2/3). A guinea-pig reference antiserum and a negative guinea-pig serum are included on each plate. Peroxidase-conjugated rabbit or goat antibody directed against guinea-pig IgG is added, followed by a peroxidase substrate. Optical density is measured and the relative antibody titre is calculated by the usual statistical methods (5.3).
Reagents and equipment:
Method
The description below is given as an example of a suitable plate layout but others may be used. Wells 1A-H are used for negative control serum. Wells 2-12 A-H are used for guinea-pig reference antiserum (usually in 2 positions) and individual sera from guinea-pigs immunised with the test or reference vaccine.
Coat each well of the ELISA plates with 100 µL of the appropriate antigen solution (PT, FHA and Fim 2/3 at 2 µg/mL and PRT at 4 µg/mL, in carbonate coating buffer pH 9.6). Allow to stand overnight at 4 °C in a humid atmosphere. To avoid temperature gradient effects, do not stack more than 4 plates high. On the following day, wash the plates thoroughly with the washing buffer. Block the plates by addition of 150 µL of the diluent block buffer to each well. Incubate in a humid atmosphere at 37 °C for 1 h. Wash the plates thoroughly with the washing buffer. Place 100 µL of the diluent block buffer in each well of the plates, except those of row A. Prepare suitable dilutions of individual test and reference vaccine serum samples, reference antiserum and negative control serum samples. Allocate the negative control serum to column 1, the reference antiserum to at least 2 other columns and individual test and reference vaccine sera to the remaining columns and add 100 µL of each serum to the first 2 wells of the column to which it is allocated. Using a multichannel micropipette, make 2-fold serial dilutions from row B down the plate to row H, by transferring 100 µL from one well to the next. Discard 100 µL from the last row so that all wells contain 100 µL. Incubate at 37 °C for 2 h. Wash thoroughly with the washing buffer. Prepare a suitable dilution of the peroxidase conjugate in the diluent block buffer and add 100 µL to each well. Incubate at 37 °C in a humid atmosphere for 1 h. Wash the plates thoroughly with the washing buffer. Add 100 µL of the peroxidase substrate to each well. Allow to stand at room temperature, protected from light, for 30 min. Read the plates at 405 nm in the same order as the addition of substrate was made.
7. In vivo assay of poliomyelitis vaccine (inactivated)
The capacity of the vaccine to induce the formation of neutralising antibodies is determined in vivo by one of the following methods.
TEST IN CHICKS OR GUINEA-PIGS
Prepare a suitable series of not fewer than 3 dilutions of the vaccine to be examined using a suitable buffered saline solution. Distribute either guinea-pigs weighing 250-350 g or 3-week-old chicks into groups of 10, and allocate a group to each dilution of the vaccine. Inject intramuscularly into each animal 0.5 mL of the dilution intended for its group. Bleed the animals after 5-6 days and separate the sera. Examine the sera for the presence of neutralising antibodies, at a dilution of 1 in 4, to each of the human poliovirus types 1, 2 and 3. Mix 100 CCID50 of virus with the dilution of serum and incubate at 37 °C for 4.5-6 h. Keep at 5 ± 3 °C for 12-18 h where necessary for consistency of results. Inoculate the mixtures into cell cultures for the detection of unneutralised virus and read the results up to 7 days after inoculation. For each group of animals, note the number of sera that have neutralising antibodies and calculate the dilution of the vaccine that gives an antibody response in 50 per cent of the animals. Carry out in parallel a control test using a suitable reference preparation. The vaccine complies with the test if a dilution of 1 to 100 or more produces an antibody response for each of the 3 types of virus in 50 per cent of the animals.
TEST IN RATS
A suitable in vivo assay method consists of intramuscular injection into the hind limb(s) of not fewer than 3 dilutions of the vaccine to be examined and a reference vaccine, using for each dilution a group of 10 specific pathogen-free rats of a suitable strain. Use of 4 dilutions is often necessary to obtain valid results for all 3 serotypes. The number of animals per group must be sufficient to obtain results that meet the validity criteria; groups of 10 rats are usually sufficient, although valid results may be obtained with fewer animals per group. If animals of different sex are used, males and females are evenly distributed between all groups. A weight range of 175-250 g has been found to be suitable. An inoculum of 0.5 mL per rat is used. The dose range is chosen such that a dose response to all 3 poliovirus types is obtained. Bleed the animals after 20-22 days. Neutralising titres against all 3 poliovirus types are measured separately using 100 CCID50 of the Sabin strains as challenge viruses, Vero or Hep2 as indicator cells, and neutralisation conditions of 3 h at 35-37 °C followed by 18 h at 2-8 °C where necessary for consistency of results. Results are read following fixation and staining after 7 days of incubation at 35 °C. For a valid antibody assay, the titre of each challenge virus must be shown to be within the range 10 CCID50 to 1000 CCID50 and the neutralising antibody titre of a control serum must be within 2 twofold dilutions of the geometric mean titre of the serum. The potency is calculated by comparison of the proportion of responders for the vaccine to be examined and the reference vaccine by the probit method or, after validation, using a parallel-line model. For the probit method it is necessary to establish a cut-off neutralising antibody titre for each poliovirus type to define a responder. Due to interlaboratory variation, it is not possible to define cut-off values that could be applied by all laboratories. Rather, the cut-off values are determined for each laboratory based on a minimum series of 3 tests with the reference vaccine. The mid-point on a log2 scale of the minimum and maximum geometric mean titres of the series of 3 or more tests is used as the cut-off value. For each of the 3 poliovirus types, the potency of the vaccine is not significantly less than that of the reference preparation. The test is not valid unless:
The following section is published for information.
Guideline on waiving of the in vivo assay of poliomyelitis vaccine (inactivated) and its combinations
This guideline applies to vaccines derived from wild strains of poliovirus. The validation described should be carried out for each product before waiving of the in vivo assay, and should be repeated wherever there is a substantial change to the manufacturing process that may affect the in vitro or in vivo assays.
The European convention on the protection of vertebrate animals used for experimental and other scientific purposes requires that tests in animals shall not be carried out if a scientifically satisfactory alternative is reasonably and practically available. The aim of this guideline is therefore to promote waiving of the in vivo assay wherever it can be shown for a given product that the in vitro assay (D-antigen determination) gives sufficient assurance of satisfactory potency for routine batch control.
For the in vivo assay, the test in rats is considered to be the method of choice. For vaccines that are assayed using chicks or guinea-pigs and that have an established record of production history, the in vivo assay may be waived if the rat assay is also applied to the batches included in the validation study described below. For vaccines not yet approved, the results of the rat assay on all final bulks should be included in all data generated for demonstration of consistency of production before waiving of the in vivo assay.
Once the in vivo assay has been waived, batches of vaccine will be released on the basis of the in vitro assay, and the in vivo assay should not be used as an alternative for the release of a batch that fails the in vitro assay. Repetition of the in vitro assay may be performed according to an authorised procedure.
PROCEDURE
The following conditions should be met before performance of the validation study:
The validation study should be performed on:
These batches are assayed using as reference standard a homologous production batch:
Waiving of the in vivo assay is acceptable if the representative final bulk/lot complies with the in vivo and in vitro assays and the sub-potent batches fail to comply. If a sub-potent batch fails to comply with the D-antigen assay but complies with the in vivo assay, the latter may be repeated.
8. Flocculation value (Lf) of diphtheria and tetanus toxins and toxoids (Ramon assay)
The content of toxin or toxoid in a sample can be expressed as a flocculation value (Lf) using the Ramon assay. In this assay, antitoxin is added in increasing concentrations to a series of tubes containing a constant amount of toxin or toxoid. At the equivalence point of toxin/toxoid and antitoxin, flocculation occurs in 1 or more tubes. The first tube in which flocculation occurs is used to determine the Lf value of the sample.
The Lf value of a toxin or toxoid is determined by the number of units of antitoxin that, when mixed with the sample, produces an optimally flocculating mixture (Ramon assay).
Practical experience has shown that the results of the calibration of antitoxins in International Units (IU), for example by comparison to international antitoxin standards, depends on the immunochemical method used. For this reason, antitoxins used for the Ramon assay must be directly calibrated against the international biological reference reagents for diphtheria or tetanus toxoid for flocculation tests, using the principles described below. The concentration thus determined may be indicated in Lf-equivalents per millilitre (Lf-eq./mL).
By definition, 1 Lf is the quantity of toxin or toxoid that flocculates in the shortest time with 1 Lf-eq. of specific antitoxin.
A range of volumes of the reference standard of antitoxin adjusted to a concentration of 100 Lf-eq./mL is dispensed into a series of, for example, 7 cm × 1 cm flocculation tubes. A sufficient quantity of a 9 g/L solution of sodium chloride R is added to each tube to give a constant total volume of, for example, 1 mL. The test sample is diluted to give an expected concentration of approximately 50 Lf/mL, and, for example, 1 mL aliquots of this dilution are dispensed into each of the tubes containing antitoxin. The tubes are properly mixed by shaking, then placed in a water-bath at a constant temperature between 30 °C and 52 °C, and observed at regular intervals for the first appearance of floccules. This may require the use of a magnifying lens and strong illumination.
The first and the second mixtures to flocculate are recorded as well as the time taken for the first flocculation to appear. 2 tubes may flocculate simultaneously.
The first tube to flocculate is the one that contains the amount of antitoxin closest in equivalence to the amount of antigen in the sample. The antitoxin content of this tube can be used to calculate the Lf value of the sample. If 2 tubes flocculate at the same time, the mean from the tubes are given as the result.
The time taken for the first tube to flocculate (Kf) is a useful indicator of the quality of the antigen. If at a given temperature and concentration of toxoid and antitoxin the Kf value is increased compared with normal, this indicates that the antigen has been damaged. The Kf value may also change with the quality of the antitoxin used.
Example
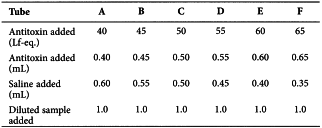
If in this example the first tube to flocculate is tube C then the Lf value of the diluted sample is 50 Lf/mL. However, if the first tube to flocculate is tube A or tube F this does not indicate equivalence at that level. It would be necessary to perform a repeat test using either a different dilution of test sample or selecting a different range of doses of reference antitoxin.
More precision can be obtained by making allowance for the sequence of flocculation after the first tube. Thus, in the example quoted, if the second tube to flocculate had been tube D, the final value for the diluted sample would be 52, whereas if the second tube to flocculate was tube B, the final value would be 48. The test may be performed in duplicate with slightly different dilutions of the test sample.
If there is no indication of the expected Lf value of the sample available, it is advisable to obtain a rough estimate by use of a wider range of antitoxin content in the tubes before proceeding to the final test.
Example
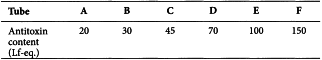
The level of toxin or toxoid and antitoxin concentration in the test may be varied, but this will markedly affect the flocculation time, so that at very low levels the test will take too long, whilst at a high concentration the onset of flocculation may be so rapid as to make it difficult to distinguish the first and second tubes to flocculate.
Assay of low concentrations by blend flocculation
For very low concentrations, it is preferable to measure toxin or toxoid by the method of blend flocculation. This involves comparison of the Lf value of a known toxin or toxoid and that of a mixture of the sample with that toxin or toxoid.
When a toxin or toxoid with a known Lf value and a toxin or toxoid with an unknown Lf value are flocculated together, the mixture will flocculate as the sum of their values if they are homogeneous. If non-homogenous toxins or toxoids are mixed they will produce an aberrant pattern with 2 flocculation maxima.
9. Residual pertussis toxin and irreversibility of pertussis toxoid
This test is not necessary for the product obtained by genetic modification.
Pertussis toxin BRP or an in-house toxin preparation calibrated in International Units against the BRP are suitable for use as a reference pertussis toxin preparation.
Mouse strain A suitable mouse strain has a toxin LD50 between 6 IU and 50 IU.
Use equal groups of not fewer than 10 histamine-sensitive mice of suitable strain, age and weight. For the test for residual pertussis toxin, inject intraperitoneally into the 1st group between 1 and 2 human doses of the vaccine stored at 2-8 °C. For the test for irreversibility of pertussis toxoid, inject intraperitoneally into the 2nd group between 1 and 2 human doses of the vaccine incubated at 37 °C for 4 weeks.
The histamine sensitivity criteria must be set during a validation study using multiple doses of a reference toxin; a significant dose-response must be demonstrated. A suitable dose of the reference toxin, chosen in the linear region of the dose-response curve and giving a positive response considered appropriate by the competent authority, is subsequently included in each assay as the positive control to demonstrate assay sensitivity.
The positive control group of mice is injected with an equivalent volume of reference pertussis toxin preparation (e.g. pertussis toxin BRP) at a dose that has been defined in the validation stage as demonstrating the assay sensitivity, for example a dose causing death in at least 30 per cent of the mice.
The negative control group of mice is injected intraperitoneally with an equivalent volume of diluent.
If a reference group of mice is used, it may be injected with a reference pertussis toxin preparation (e.g. pertussis toxin BRP) at a dose previously set as the allowable upper limit of pertussis toxin in the product according to historical safety data. Alternatively, a reference vaccine with established clinical safety may be used instead of the reference toxin preparation.
After 5 days, inject intraperitoneally into each mouse of each group the equivalent of 2 mg of histamine base in a volume not exceeding 0.5 mL and observe for 24 h. The test is not valid if:
If a reference group is not included in the assay, the vaccine complies with the test for residual pertussis toxin if no mice die in the 1st group. If a vaccine lot fails the test, the test may be repeated once with the same number of mice or with a greater number and the results of the tests combined; the vaccine complies with the test if the percentage of deaths in the group given the vaccine does not exceed 5 per cent. The vaccine complies with the test for irreversibility of pertussis toxoid if the 2nd group, given the vaccine incubated at 37 °C, also complies with these criteria.
If a reference group is included, the vaccine complies with the test for residual pertussis toxin if the percentage of deaths in the 1st group is not greater than that in the reference group. If a vaccine lot fails the test, the test may be repeated once with the same number of mice or with a greater number and the results of the tests combined; the vaccine complies with the test if the percentage of deaths in the groups given the vaccine does not exceed the percentage of deaths in the reference groups. The vaccine complies with the test for irreversibility of pertussis toxoid if the 2nd group, given the vaccine incubated at 37 °C, also complies with these criteria.
Alternatively, after validation, a histamine-sensitisation test based on body temperature measurements as end-points may be used instead of the lethal end-point test in mice.