SC I Guidelines for using the test for bacterial endotoxins
1. Introduction
Endotoxins from gram-negative bacteria are the most common cause of toxic reactions resulting from contamination of pharmaceutical products with pyrogens; their common pyrogenic activity is much higher than that of other known pyrogenic substances. These endotoxins are lipopolysaccharides. Although there are a small number of pyrogens that possess a different structure, the conclusion is generally justified that the absence of bacterial endotoxins in a substance or product implies the absence of pyrogenic components, provided the presence of non-endotoxin pyrogenic substances can be ruled out. The monocyte-activation test (2.6.30) is a suitable method to use to rule out the presence of non-endotoxin pyrogens in substances or products.
The presence of endotoxins in a substance or product may be masked by factors interfering with the reaction between the endotoxins, the test reagents and the amoebocyte lysate. Also, the ability to detect endotoxins may be affected by storage conditions or storage time. Hence, the analyst who wishes to implement a test for bacterial endotoxins or to replace the pyrogen test by a test for bacterial endotoxins has to demonstrate that a valid test can be carried out on the substance or product concerned; this may entail a procedure for removing interference.
As indicated in general chapter 2.6.14. Bacterial endotoxins, information must be available on the following 2 aspects before a test on a sample can be regarded as valid.
General chapter 2.6.14. Bacterial endotoxins indicates methods for removing interfering factors; in the case of interference, another test must be carried out after such a method has been applied to check whether the interference has indeed been neutralised or removed.
This general chapter explains the reasons for the requirements in the test for bacterial endotoxins, then deals with reading and interpretation of the results.
Replacement of the rabbit pyrogen test required in a pharmacopoeial monograph by an amoebocyte lysate test, or by other methods such as the monocyte-activation test or a test using recombinant factor C reagent as a replacement for the amoebocyte lysate, constitutes the use of an alternative method of analysis and hence requires demonstration that the method is appropriate for the given substance or product and gives a result consistent with that obtained with the prescribed method as described in the General Notices (see also section 12).
The prescribed method for bacterial endotoxins may be stated in the monograph on a given substance or product. The use of a method other than the method prescribed in the monograph is considered as the use of an alternative method. Where no method is stated, any of methods A to F of general chapter 2.6.14. Bacterial endotoxins can be used.
2. Method AND ACCEPTANCE CRITERIA
2-1 Methods and precautions to be taken
The addition of endotoxins to amoebocyte lysate may result in turbidity, precipitation or gelation (gel-clot); initially only the gel-clot method was used in the Pharmacopoeia as an evaluation criterion in the test for bacterial endotoxins. The advantage was the simplicity of basing the decision to pass or fail the substance or product to be examined on the absence or presence of a gel-clot, visible with the naked eye. The quantitative methods C, D, E and F were developed later: they require more instrumentation, but they are easier to automate for the regular testing of large numbers of samples of the same substance or product.
Endotoxins may be adsorbed onto the surface of tubes or pipettes made from certain plastics or types of glass. Interference may appear due to the release of substances from plastic materials. Hence, the materials used must be checked.
2-2 Endotoxin limit concentration
The decision to use the test for bacterial endotoxins as a limit test implies firstly that an endotoxin limit concentration must be defined for the substance or product to be examined, and secondly that the objective of the test is to know whether the endotoxin concentration in the sample to be examined is below or above this limit. The quantitative methods C, D, E and F make it possible to determine the endotoxin concentration in the sample to be examined, but for compliance with the Pharmacopoeia and in routine quality control the final question is whether or not this concentration exceeds a defined limit.
The dose of the substance or product to be examined must be taken into account in setting the endotoxin limit concentration: the limit is set so as to ensure that, as long as the endotoxin concentration in the substance or product remains below this limit, even the maximal dose administered by the intended route per hour does not contain sufficient endotoxin to cause a toxic reaction.
When the endotoxin concentration in the substance or product exactly equals the endotoxin limit concentration, gelation will occur, as is the case when the endotoxin concentration is much higher, and the substance or product will fail the test, because the all-or-none character of the test makes it impossible to differentiate between a concentration exactly equal to the endotoxin limit concentration and one that is higher. It is only when no gelation occurs that the analyst may conclude that the endotoxin concentration is below the endotoxin limit.
For substances or products in the solid state, this endotoxin limit concentration per mass unit or per International Unit (IU) of substance or product has to be converted into a concentration of endotoxin per millilitre of solution to be examined, as the test can only be carried out on a solution. The case of substances or products that already exist in the liquid state (such as infusion fluids) is discussed below.
2-3 Calculation of the endotoxin limit
The endotoxin limit for active substances administered parenterally, defined on the basis of dose, is equal to:
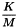
| K | = | threshold pyrogenic dose of endotoxin per kilogram of body mass; |
| M | = | maximum recommended bolus dose of product per kilogram of body mass. |
When the product is to be injected at frequent intervals or infused continuously, M is the maximum total dose administered per hour.
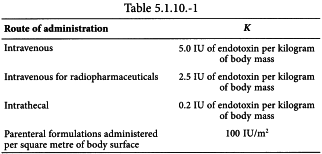
The endotoxin limit depends on the product and its route of administration and may be stated in the monograph. Values for K are suggested in Table 5.1.10.-1.
For other routes, the acceptance criterion for bacterial endotoxins is generally determined on the basis of results obtained during the development of the preparation.
2-4 Considerations when establishing an endotoxin limit for a specific substance or product
The endotoxin limit for a substance or product is established with consideration of the following aspects.
Calculated endotoxin limit
The endotoxin limit is calculated as described in section 2-3. This represents a safety limit not to be exceeded if the product is to be administered to humans.
Limit prescribed in an individual substance monograph
The limit stated in an individual substance monograph frequently reflects what is achievable in a controlled production environment. The limit prescribed in a monograph can therefore be lower than the calculated endotoxin limit. However a manufacturer may specify a limit that is more stringent than that stated in the monograph.
Process capability
The capability of the process to reduce or remove bacterial endotoxins during manufacture might result in lower endotoxin limits for specific processes.
Additional safety requirements
Precautions are taken in consideration of patient population (such as paediatric use, malnourished or cachectic patients, etc.), specific local requirements (e.g. countries might wish to operate with a lower average body weight of 60 kg instead of 70 kg frequently employed in Europe) or any additional safety margins requested by the competent authority.
Formulation of the product
The limit must take into consideration any theoretical bacterial endotoxin load introduced by any other components used for reconstitution and/or dilution of the product (e.g. water for injections) or introduced by starting materials and/or raw materials.
2-5 Maximum Valid Dilution
Which dilution of the substance or product is to be used in the test to obtain maximal assurance that a negative result means that the endotoxin concentration of the substance or product is less than the endotoxin limit and that a positive result means that the lysate detected an endotoxin concentration equal to or greater than the endotoxin limit? This dilution depends on the endotoxin limit and on the sensitivity of the lysate; it is called the maximum valid dilution (MVD) and its value may be calculated using the following expression:
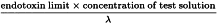
| λ | = | the labelled lysate sensitivity in the gel-clot technique (IU/mL) or the lowest concentration used in the standard curve of the turbidimetric or chromogenic techniques. |
When the value of the MVD is not a whole number, a convenient whole number smaller than the MVD may be used for routine purposes (which means preparing a solution of the substance or product that is less diluted than the MVD indicates). In this case, a negative result indicates that the endotoxin concentration of the substance or product lies below the limit value. However, when the endotoxin concentration of the substance or product in such a test is less than the endotoxin limit but high enough to make the reaction with the lysate result in a clot, the test may be positive under these conditions. Hence, when a test with this ‘convenient’ dilution factor is positive, the substance or product is diluted to the MVD and the test is repeated. In any case of doubt or dispute, the MVD must be used.
This stresses the importance of the confirmation of the sensitivity of the lysate.
Example
A 50 mg/mL solution of phenytoin sodium (intended for intravenous injection) has to be tested. Determine the MVD, given the following variables:
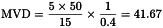
| M | = | maximum human dose = 15 mg per kilogram of body mass; |
| c | = | 50 mg/mL; |
| K | = | 5 IU of endotoxin per kilogram of body mass; |
| λ | = | 0.4 IU of endotoxin per millilitre. |
For routine tests on this product, it may be expedient to dilute 1 mL of the solution to be examined to 20 mL (MVD/2 rounded to the next lower whole number). However, if this test result is positive the analyst will have to dilute 1 mL to 41.67 mL and repeat the test. A dilution to 41.67 mL is also necessary when the test is performed to settle a dispute.
3. RISK ASSESSMENT
As stated in section 1 of this general chapter, the conclusion is generally justified that the absence of bacterial endotoxins in a substance or product implies the absence of pyrogenic components, provided the presence of non-endotoxin pyrogenic substances can be ruled out. To rule out the presence of non-endotoxin pyrogens in substances or products, the use of the monocyte-activation test (2.6.30) is recommended at release or during development of the production process; if any changes are made to the production process that could influence the quality of the product regarding pyrogenicity, the monocyte-activation test is repeated. Examples of such changes include the use of different raw materials, a different production site and different process parameters.
The decision to use the test for bacterial endotoxins as the sole pyrogenicity test is to be made after careful evaluation of the risk of the substance or product containing non-endotoxin pyrogens. The risk assessment is made with consideration given to any factor that could result in the inclusion of pyrogens not detected by the test for bacterial endotoxins. The items below constitute a non-exhaustive list of factors to be considered in the risk assessment.
Production process
(chemical synthesis, fermentation, biotechnological method). For products of fermentation, the expression system is to be considered (prokaryotic, eukaryotic) and, for a prokaryotic expression system, whether gram-positive or gram-negative bacteria are used. Also, the culture media components are examined with consideration given to their origin (synthetic, animal, plant).
Bioburden
The potential presence of gram-positive bacteria and fungi as contaminants of the active substance, excipients or starting materials and raw materials used in the production of the medicinal product, and the origin of the raw materials (synthetic, animal, plant) have to be taken into consideration. The quality of the water plays an important role on the overall evaluation.
Capability of the downstream process
It must be verified whether bacterial endotoxin removal steps are part of the downstream process.
Safety
The target population and the route of administration (e.g. intravenous, intrathecal) have to be taken into account in the risk assessment.
Stability of the detectable endotoxins
It has to be considered that the ability to detect endotoxins can be affected by interaction with certain components, storage conditions or storage time, temperature and handling of the test sample. Procedures that demonstrate stability of the detectable endotoxin content have to be established for storing, handling and mixing of samples.
4. Reference material
Endotoxin standard BRP is intended for use as the reference preparation. It has been assayed against the WHO International Standard for Endotoxin and its potency is expressed in International Units of endotoxin per vial. The International Unit of endotoxin is defined as the specific activity of a defined mass of the International Standard.
For routine purposes, another preparation of endotoxin may be used, provided it has been assayed against the International Standard for Endotoxin or the BRP and its potency is expressed in International Units of endotoxin.
NOTE: 1 International Unit (IU) of endotoxin is equal to 1 Endotoxin Unit (E.U.).
5. Water for bet
Water for BET is sterile water that is free of detectable levels of endotoxin. Usually it is commercially available and certified.
General chapter 2.6.14. Bacterial endotoxins indicates that methods other than triple distillation may be used to prepare water for BET. Reverse osmosis has been used with good results; some analysts may prefer to distil the water more than 3 times. Whatever method is used, the resultant product must be free of detectable bacterial endotoxins.
6. pH OF THE MIXTURE
In the test for bacterial endotoxins, optimum gel-clot occurs for a mixture at pH 6.0-8.0. However, the addition of the lysate to the sample may result in a lowering of the pH.
7. Validation of the lysate
It is important to follow the manufacturer’s instructions for the preparation of the solutions of the lysate.
The positive end-point dilution factors in gel-clot methods A and B are converted to logarithms. The reason is that if the frequency distribution of these logarithmic values is plotted, it usually approaches a normal distribution curve much more closely than the frequency distribution of the dilution factors themselves; in fact it is so similar that it is acceptable to use the normal frequency distribution as a mathematical model and to calculate confidence limits with Student’s t-test.
8. Preliminary test for interfering factors
Some substances or products cannot be tested directly for the presence of bacterial endotoxins because they are not miscible with the reagents, they cannot be adjusted to pH 6.0-8.0 or they inhibit or activate enzymatic reaction (such as β-D-glucans).
Therefore a preliminary test is required to check for the presence of interfering factors; when these are found the analyst must demonstrate that the procedure to remove them has been effective and that by applying this procedure, any bacterial endotoxins present have not been removed.
The object of the preliminary test is to test the null hypothesis that the sensitivity of the lysate in the presence of the substance or product to be examined does not differ significantly from the sensitivity of the lysate in the absence of the product. A simple criterion is used in methods A and B: the null hypothesis is accepted when the sensitivity of the lysate in the presence of the product is at least 0.5 times and not more than twice the sensitivity of the lysate by itself.
The test for interfering factors in gel-clot methods A and B requires the use of a sample of the substance or product in which no endotoxins are detectable. This presents a theoretical problem when an entirely new product has to be tested. Hence, a different approach was designed for quantitative methods C, D, E and F.
Note that methods D and E, which use a chromogenic peptide, require reagents that are absent in methods A, B, C and F, and hence compliance of methods A, B, C or F with the requirements for interfering factors cannot be extrapolated to method D or method E without further testing.
9. Removal of interfering factors
The procedures to remove interfering factors must not increase or decrease (for example, by adsorption) the amount of endotoxin in the substance or product to be examined. The correct way of checking this is to apply the procedures to a spiked sample of the substance or product to be examined, that is, a sample to which a known amount of endotoxin has been added, and then to measure the recovery of the endotoxin after the removal process has been conducted.
Methods C and D
If the nature of the product to be examined results in an interference that cannot be removed by classical methods (e.g. dilution or centrifugation), it may be possible to determine the standard curve in the same type of substance or product freed from endotoxins by appropriate treatment or by dilution of the substance or product. The endotoxins test is then carried out by comparison with this standard curve.
Ultrafiltration with cellulose triacetate asymmetric membrane filters has been found to be suitable in most cases. The filters must be properly validated, because under some circumstances cellulose derivatives (β-D-glucans) can cause false positive results.
Another option to remove interfering factors is a 2-step procedure in which 1) endotoxin within the interfering sample is fixed on a solid phase, and 2) after removal of the interfering substance (e.g. by washing) the endotoxin is detected unimpaired under suitable testing conditions.
10. The purpose of the controls
The purpose of the control made up with water for BET and the reference preparation of endotoxin at twice the concentration of the labelled lysate sensitivity is to verify the activity of the lysate at the time and under the conditions of the test (for method A and B). The purpose of the negative control is to verify the absence of a detectable concentration of endotoxin in the water for BET.
The positive control, which contains the product to be examined at the concentration used in the test, is intended to show the absence of inhibiting factors at the time and under the conditions of the test.
11. READING AND INTERPRETATION OF RESULTS
Minute amounts of bacterial endotoxin in the water for BET, or in any other reagent or material to which the lysate is exposed during the test, may escape detection as long as they do not reach the sensitivity limit of the lysate. However, they may raise the amount of bacterial endotoxin in the solution containing the substance or product to be examined to just above the sensitivity limit and cause a positive reaction.
The risk of this happening may be reduced by testing the water for BET and the other reagents and materials with the most sensitive lysate available, or at least one that is more sensitive than the one used in the test on the product. Even then, the risk of such a ‘false positive result′ cannot be ruled out completely.
12. REPLACEMENT OF METHODS prescribed IN MONOGRAPHS
12-1 Replacement by another Ph. Eur. method
As stated in the General Notices, the test methods given in monographs and general chapters have been validated in accordance with accepted scientific practice and current recommendations on analytical validation. The methods described in general chapters 2.6.14. Bacterial endotoxins and 2.6.30. Monocyte-activation test therefore do not have to be re-validated per se, other than in consideration of their use for a specific substance or product in a specific analytical environment.
The procedure and the materials and reagents used in the method must be validated as described for the test concerned.
The absence of interfering factors (and, if necessary, the procedure for removing them) is verified on samples of at least 3 production batches.
The necessary information is sought from manufacturers; companies are invited to provide any validation data that they have concerning the applicability of the replacement test to the substances and products of interest; such data includes details of sample preparation and of any procedures necessary to eliminate interfering factors.
As stated in general chapter 2.6.30. Monocyte-activation test, the monocyte-activation test is primarily intended as a replacement of the rabbit pyrogen test. Guidelines on which methods to use (A, B or C) and on how to validate the monocyte-activation test are described in general chapter 2.6.30. Monocyte-activation test.
12-2 Replacement by an alternative method not described in the Ph. Eur.
The use of alternative reagents such as recombinant factor C as a replacement to the amoebocyte lysate eliminates the use of a reagent extracted from live animals.
Replacement of a rabbit pyrogen test or a bacterial endotoxin test prescribed in a monograph by a test using recombinant factor C reagent or any other reagent as a replacement of the amoebocyte lysate is to be regarded as the use of an alternative method in the replacement of a pharmacopoeial test, as described in the General Notices.