Appendix XIV H. Monocyte-Activation Test
1 Introduction
The monocyte-activation test (MAT) is used to detect or quantify substances that activate human monocytes or monocytic cells to release endogenous mediators such as pro-inflammatory cytokines, for example tumour necrosis factor alpha (TNFα), interleukin-1 beta (IL-1β) and interleukin-6 (IL-6). These cytokines have a role in fever pathogenesis. Consequently, the MAT will detect the presence of pyrogens in the test sample. The MAT is suitable, after a product-specific validation, as a replacement for the rabbit pyrogen test.
Pharmaceutical products that contain non-endotoxin pyrogenic or pro-inflammatory contaminants often show very steep or non-linear dose-response curves in comparison with endotoxin dose-response curves. Preparations that contain or may contain non-endotoxin contaminants have to be tested at a range of dilutions that includes minimum dilution.
The following 3 methods are described in the present chapter.
Method A. Quantitative test
Method B. Semi-quantitative test
Method C. Reference lot comparison test
In addition, further useful information on practical aspects of the tests can be found in the ‘Guidance notes’ section at the end of this general chapter.
The test is carried out in a manner that avoids pyrogen contamination.
2 Definitions
The maximum valid dilution (MVD) is the maximum allowable dilution of a sample at which the contaminant limit can be determined. The calculation of the MVD is based on the endotoxin reference standard. Determine the MVD using the following expression:

| CLC | = | contaminant limit concentration; |
| C | = | concentration of test solution; |
| LOD | = | limit of detection. |
As the LOD is not always available in advance, an estimated LOD based on historical data can be used to calculate the MVD.
The acceptance criterion for a pass/fail decision is the contaminant limit concentration (CLC), which is expressed in endotoxin equivalents per milligram or millilitre or per units of biological activity of the preparation being examined.
The CLC is calculated using the following expression:
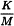
| K | = | threshold pyrogenic dose per kilogram of body mass; |
| M | = | maximum recommended bolus dose of product per kilogram of body mass. |
When the product is to be injected at frequent intervals or infused continuously, M is the maximum total dose administered in a single hour period.
Where an endotoxin limit concentration (ELC) has been specified for a product, the CLC is the same as the ELC, unless otherwise prescribed. In this case, the concentration of test solution is expressed in mg/mL if the endotoxin limit is specified by mass (IU/mg), in Units/mL if the endotoxin limit is specified by unit of biological activity (IU/Unit), in mL/mL if the endotoxin limit is specified by volume (IU/mL).
Endotoxin equivalents are values for the contaminant concentration read off the standard endotoxin dose-response curve (Method A) or estimated by comparison with responses to standard endotoxin solutions (Method B). The standard endotoxin stock solution is prepared from an endotoxin reference standard that has been calibrated against the International Standard, for example endotoxin standard BRP.
The LOD is determined using the endotoxin standard curve. It is the concentration of endotoxin corresponding to the cut-off value. For the purpose of the test, the LOD is expressed as endotoxin equivalents per millilitre. The cut-off value is expressed in units appropriate to the read-out (e.g. for an enzyme-linked immunosorbent assay (ELISA), use the optical density).
The cut-off value may be calculated using the following expression:
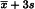
| x̄ | = | mean of the 4 replicates for the responses to the blank (R0); |
| s | = | standard deviation of the 4 replicates of the responses to the blank (R0). |
3 General procedure
A solution of the preparation being examined is incubated with a source of human monocytes or human monocytic cells, e.g. from human heparinised peripheral blood that is preferably not more than 4 h old, or a monocyte-containing fraction of that blood, such as human peripheral blood mononuclear cells (PBMC) isolated, e.g. by density-gradient centrifugation, or a human monocytic cell line. Human heparinised peripheral blood is usually diluted with culture medium or saline e.g. to 2-50 per cent V/V (final concentration). PBMC or monocytic cell lines, in culture medium and with either the donor′s own plasma or AB serum, are typically used at a final cell density of 0.1-1.0 × 106 cells per well, tube or other receptacle. For monocytic cell lines, heat-inactivated foetal bovine serum may be substituted for AB serum. The cell culture is carried out at 37 ± 1 °C, in an atmosphere appropriate for the culture medium, e.g. 5 per cent CO2 in humidified air. The duration of the culture is sufficient to allow accumulation of the chosen read-out. The responses of the chosen read-out, e.g. a pro-inflammatory or pyrogenic cytokine, to a solution of the preparation being examined are compared with responses to standard endotoxin or to a reference lot of the preparation being examined.
4 Apparatus
Depyrogenate all glassware and other heat-stable apparatus in a hot-air oven using a validated process. A commonly used minimum time and temperature is 30 min at 250 °C. If employing plastic apparatus, such as microtitre plates and pipette tips for automatic pipetters, use apparatus shown to be free of detectable pyrogens and which do not interfere with the test.
5 CELL SOURCES AND QUALIFICATION
5-1 Whole blood
Whole blood is obtained from single donors or from pooled whole blood which are qualified according to the requirements described under sections 5-3, 5-4, 5-5 and where applicable, section 6-3.
5-2 Peripheral blood mononuclear cells (PBMC)
PBMC are isolated from blood obtained from single donors or from pooled whole blood which are qualified according to the requirements described under sections 5-3, 5-4, 5-5 and where applicable, section 6-3.
5-3 Qualification of blood donors
Blood donors are to satisfy the following qualification criteria, together with other requirements in force that relate to consent, health and safety and ethical considerations. Blood donors are to describe themselves as being in good health, as not to be suffering from any bacterial or viral infections and to have been free from the symptoms of any such infection for a period of at least 1 week prior to the donation of blood. Blood donors are not to have taken non-steroidal anti-inflammatory drugs during the 48 h prior to donating blood and steroidal anti-inflammatory drugs during the 7 days prior to donating blood. Individuals who have been prescribed immunosuppressant or other drugs known to influence the production of the chosen readout are not to serve as blood donors. Blood donations are to be tested for infection markers according to national requirements for transfusion medicine.
5-4 Qualification of cells pooled from a number of donors
Pools (of whole blood or blood fractions, e.g. PBMC), must consist of donations from a minimum of 4 individual donors but preferably 8 or more donors, where practicable, taking from each donation an approximately equal volume of blood, or cells from an approximately equal volume of blood. For the qualification of pooled cells proceed as follows: within 4 h of collection of blood, generate dose-response curves from the pool using standard endotoxin with at least 4 geometrically diluted endotoxin concentrations, e.g. in the range of 0.01 IU/mL to 4 IU/mL. The dose-response curves are to meet the 2 criteria for the endotoxin standard curve described under section 6-1. If the pool is to be used for the detection of non-endotoxin contaminants, pools are to be qualified as described in section 6-3. Where cells are pooled, the averaging effect is to be considered when setting the pass/fail specification for a given product.
5-5 Qualification of cryo-preserved cells
The cell source intended for use in a MAT, e.g. human whole blood, blood fractions, such as PBMC or monocytic cell lines, may be cryo-preserved. Pools of cryo-preserved cells are obtained by pooling before freezing, or by pooling single cryo-preserved donations immediately after thawing. Qualification of cryo-preserved blood or cells is performed immediately after thawing (and pooling if necessary). Dose-response curves for the cryo-preserved blood or cells are to comply with the 2 criteria for the endotoxin standard curve described under section 6-1. Qualification of the cryo-preserved blood or cells is to be performed according to section 6-3 if the intended use is for the detection of non-endotoxin contaminants.
5-6 Monocytic continuous cell lines
Monocytic cell lines are appropriate for the detection of bacterial endotoxins, but have limited use for the detection of non-endotoxin pyrogens.
A human monocytic cell line is cultured in order to ensure a sufficient supply for the MAT. To optimise the method, clones derived from the cell line can be used.
Cells must be maintained under aseptic conditions and regularly tested for the presence of mycoplasma contamination. Additionally, cells must be regularly checked for identity (e.g. doubling time, morphology, and function) and stability. The functional stability of a cell line is assessed by monitoring its performance in relation to the number of passages during routine testing. Criteria for functional stability are to be established and may include growth criteria, maximum signal obtained in the test, background noise and receptor expression. The receptor expression may be tested with specific ligands e.g. lipopolysaccharide (LPS) for toll-like receptor 4 (TLR4), lipoteichoic acid (LTA) for toll-like receptor 2 (TLR2), synthetic bacterial lipoprotein for TLR2-TLR1, synthetic bacterial lipoprotein for TLR2-TLR6 or flagellin.
The dose-response curves are to meet the 2 criteria for the endotoxin standard curve described under section 6-1. If the cells are to be used for the detection of non-endotoxin contaminants, they are to be qualified as described in section 6-3.
6 Preparatory testing
To ensure both the precision and validity of the test, preparatory tests are conducted, to assure that the criteria for the endotoxin standard curve are satisfied, that the solution does not interfere with the test, that the test detects endotoxins and non-endotoxins contaminants and that the solution does not interfere in the detection system.
A test for interfering factors is required when any changes are made to the experimental conditions that are likely to influence the result of the test.
6-1 Assurance of criteria for the endotoxin standard curve
Using the standard endotoxin solution, prepare at least 4 endotoxin concentrations to generate the standard curve. Perform the test using at least 4 replicates of each concentration of standard endotoxin.
The basal release of the chosen read-out (blank) in the absence of added standard endotoxin is optimised to be as low as possible (e.g. an optical density below 0.1 when using an ELISA).
There are 2 acceptance criteria for the standard curve:
6-2 Test for interfering factors
To assure the validity of the test, preparatory tests are conducted to ensure that the preparation being examined does not interfere with the test. Using an appropriate diluent, dilute the preparation being examined in geometric steps, with all dilutions not exceeding the MVD. Make the same dilutions of the preparation being examined and add endotoxin at a justified concentration. Alternatively, use a diluent containing added endotoxin at a justified concentration. In both cases, this concentration is usually equal to or near the estimated middle of the endotoxin standard curve (Method A) or twice the estimated LOD (Method B). Test these dilution series in parallel in the same experiment. Use the endotoxin standard curve to calculate the concentration of endotoxin-equivalents in each solution. Calculate the mean recovery of the added endotoxin by subtracting the mean concentration of endotoxin equivalents in the solution (if any) from that in the solution containing the added endotoxin. The test solution is considered free of interfering factors if, under the conditions of the test, the measured endotoxin equivalents in the test solution to which endotoxin is added is within 50-200 per cent of the added concentration, after subtraction of any endotoxin equivalents detected in the solution without added endotoxin. When this criterion is not met, Method C is to be preferred over Methods A and B.
In Method C, the dilutions of the test and reference lots depend on the type of analysis used to make the comparison between the two. The type of analysis is to be justified and validated for each product, and is to include assay validity criteria. In an example, a solution of the preparation being examined is tested at 3 dilutions: the highest concentration (lowest dilution) that stimulates the greatest release of the chosen read-out and the 2-fold dilutions immediately below and above the chosen dilution. Since the concentration that stimulates the greatest release of the chosen read-out may be donor-dependent as well as batch-dependent, the product-specific validation is to be performed in at least 3 independent tests, each using cells from different donors. The highest concentration (lowest dilution) that stimulates the greatest release of the chosen read-out in the majority of donors, and the 2-fold dilutions immediately below and above that dilution are deemed to be validated for further testing. If undiluted test solution stimulates the greatest release of the chosen read-out, subsequent testing is to be performed using undiluted test solution and also test solution diluted in the ratios 1:2 and 1:4 before its addition to the monocytic cells. The dilution factors for these 3 solutions are designated f1, f2 and f3.
If the pyrogen content of the product is inherently high, it may be more appropriate to carry out, for example, a parallel-line analysis on the dose-response curves for the test and reference lots. In this situation, solutions of the preparations are tested at 3 or more geometric dilutions which cover the range of the dose-response curve used for the validated analysis (see chapter 5.3. Statistical analysis).
6-3 Method validation for non-endotoxin monocyte-activating contaminants
The preparatory testing is also to show that the chosen test system detects, in addition to bacterial endotoxins, non-endotoxin pro-inflammatory or pyrogenic contaminants. The suitability of the method for the particular product has to be verified. This can be achieved using historic batches found to be contaminated with non-endotoxin contaminants that caused positive responses in the rabbit pyrogens test or adverse drug reactions in man. Where such batches are not available, the preparatory testing is to include validation of the test system using at least 2 non-endotoxin ligands for toll-like receptors, e.g. peptidoglycans, lipoteichoic acids, synthetic bacterial lipoproteins, flagellin and crude bacterial whole cell extract, at least 1 of which is to be spiked into the preparation being examined. The choice of non-endotoxin pyrogens used should reflect the most likely contaminant(s) of the preparation being examined.
6-4 Interference in the detection system
Once the optimum dilution of the solution of the preparation being examined for further testing has been identified, this dilution is tested for interference in the detection system (e.g. ELISA) for the chosen read-out. The agreement between a dilution series of the standard for the chosen read-out, in the presence and absence of the preparation being examined, is to be within, for example ± 20 per cent of the optical density.
7 Methods
7-1 Method a: quantitative test
Method A involves a comparison of the preparation being examined with a standard endotoxin dose-response curve. The contaminant concentration of the preparation being examined is to be less than the CLC to pass the test.
7-1-1 Test procedure
Using the validated test method, prepare the solutions shown in Table 2.6.30.-1 and culture 4 replicates of each solution with the qualified cells.
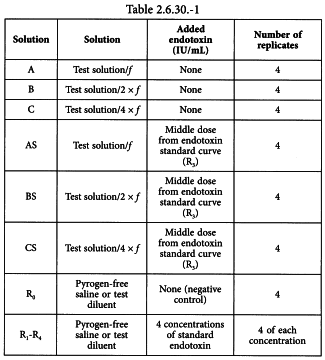
Solution A = solution of the preparation being examined at the dilution, here designated f, at which the test for interfering factors was carried out, i.e. the highest concentration (lowest dilution) for which the endotoxin recovery is within 50-200 per cent.
Solution B = 2-fold dilution of solution A, not exceeding the MVD.
Solution C = 2-fold dilution of solution B, not exceeding the MVD.
Solution AS = solution A spiked with standard endotoxin at a concentration equal to the middle dose from the endotoxin standard curve (R3).
Solution BS = solution B spiked with standard endotoxin at a concentration equal to the middle dose from the endotoxin standard curve (R3).
Solution CS = solution C spiked with standard endotoxin at a concentration equal to the middle dose from the endotoxin standard curve (R3).
Solution R0 = negative control.
Solutions R1-R4 = solutions of standard endotoxin at the concentrations used in the test for interfering factors.
7-1-2 Calculation and interpretation
All data to be included in the data analysis are to relate to cells for which the 2 criteria for the endotoxin standard curve are satisfied. For each different cell source, e.g. individual donation, donor pool, or cell line, use the endotoxin standard curve R1-R4 to calculate the concentration of endotoxin equivalents in each of the replicates of solutions A, B and C and solutions AS, BS and CS. The recovery of endotoxin equivalents calculated from the endotoxin equivalents concentration found in solutions AS, BS and CS after subtracting the endotoxin equivalents concentration found in solutions A, B and C is within the range of 50-200 per cent. Dilutions not fulfilling the spike recovery argument are not valid and are therefore excluded from further evaluation. The preparation being examined complies with the requirements of the test for a given cell source if the mean concentrations of endotoxin equivalents measured in the replicates of solutions A, B and C, after correction for dilution and concentration, are all less than the CLC specified for the preparation being examined. One valid dilution is the minimum required for a valid test.
7-1-3 Pass/fail criteria of the preparation
When cells from individual donors are used, the preparation being examined is required to comply with the test with the cells from each of 4 different donors. If the preparation being examined passes the test with cells from 3 of the 4 donors, the test is continued with cells from a further 4 donors, none of whom provided cells for the 1st test, and the preparation being examined is required to pass the test with cells from 7 of the 8 different donors (i.e. a maximum of 1 positive reaction in 8 donors is allowed). When the source of monocytes consists of cells pooled from a number of individual donors, the preparation being examined is required to pass the test with 1 pool of cells. Where a human monocytic cell line is used for the test, the preparation being examined is required to pass the test with 1 qualified passage of cells.
7-2 Method b. semi-quantitative test
Method B involves a comparison of the preparation being examined with standard endotoxin. The contaminant concentration of the preparation being examined is to be less than the CLC to pass the test. Solution A must be chosen for the pass decision, unless otherwise justified and authorised.
7-2-1 Test procedure
Using the validated test method, prepare the solutions shown in Table 2.6.30.-2 and culture 4 replicates of each solution with the qualified cells.
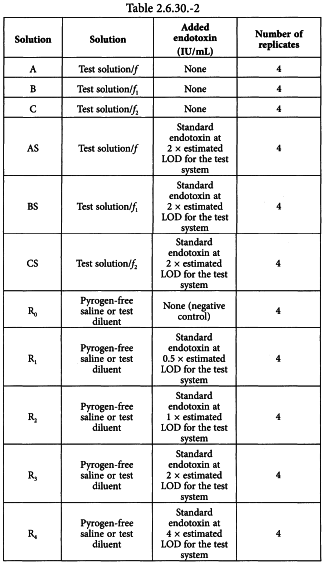
Solution A = solution of the preparation being examined at the dilution, here designated f, at which the test for interfering factors was completed.
Solution B = solution of the preparation being examined at a dilution, here designated f1, not exceeding the MVD, chosen after a review of data from the product-specific validation, e.g. 1:2 × MVD (i.e. 2 times less diluted than the MVD).
Solution C = solution of the preparation being examined at a dilution, here designated f2, not exceeding the MVD, chosen after a review of data from the product-specific validation, e.g. MVD.
Solution AS = solution A spiked with standard endotoxin at 2 × estimated LOD for the test system (as determined in preparatory testing).
Solution BS = solution B spiked with standard endotoxin at 2 × estimated LOD for the test system.
Solution CS = solution C spiked with standard endotoxin at 2 × estimated LOD for the test system.
Solution R0 = negative control.
Solution R1 = standard endotoxin at 0.5 × estimated LOD for the test system.
Solution R2 = standard endotoxin at 1 × estimated LOD for the test system.
Solution R3 = standard endotoxin at 2 × estimated LOD for the test system.
Solution R4 = standard endotoxin at 4 × estimated LOD for the test system.
7-2-2 Calculation and interpretation
All data to be included in the data analysis are to relate to cells for which mean responses to solutions R0-R4 increase progressively. The mean response to R0 may be equal to the mean response to R1. For each different cell source, the mean response to solution R2 is to be greater than a positive cut-off value. Data below this cut-off value are considered negative. If the mean response to R1 or R2 exceeds the cut-off value, the response to the solution chosen for the pass/fail decision must be negative (= pass). For each negative solution of the preparation being examined (A, B and C), the mean response to the corresponding spiked solution (AS, BS or CS respectively) is compared with the mean response to R3 to determine the percentage spike recovery. The contaminant concentration of the preparation being examined is less than the CLC for a given cell source if the solution of the preparation being examined designated for the pass/fail-decision and the dilutions below all give negative results and the endotoxin spike recovery is within the range of 50-200 per cent.
7-2-3 Pass/fail criteria of the preparation
The criteria are the same as for method A (see 7-1-3).
7-3 Method c: reference lot comparison test
Method C involves a comparison of the preparation being examined with a validated reference lot of that preparation. The type of analysis selected to compare the two is to be justified and validated for each product and is to include assay validity criteria. The reference lot is also selected according to criteria that have been justified and authorised. The test is intended to be performed in cases where a preparation being examined shows marked interference but cannot be diluted within the MVD to overcome the interference or because it contains or is believed to contain non-endotoxin contaminants. Responses to non-endotoxin contaminants may dilute out more rapidly than responses to endotoxin, which makes it necessary to perform the test at a range of dilutions that include minimum dilution. The test procedure is described below and includes an example of a type of analysis used for the comparison of a test lot and reference lot.
7-3-1 Test procedure
Using the validated test method, prepare the solutions shown in Table 2.6.30.-3 and culture 4 replicates of each solution with the qualified cells.
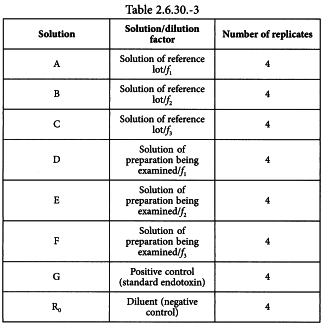
Solutions A, B and C are solutions of the reference lot diluted by the dilution factors, f1, f2 and f3, determined in the test for interfering factors.
Solutions D, E and F are solutions of the preparation being examined diluted by the dilution factors, f1, f2 and f3, determined for the reference lot in the test for interfering factors.
Solution G is the positive test control for the viability of the cells and is a standard endotoxin concentration that gives a clear positive response.
Solution R0 is the diluent used to dilute the preparation being examined and serves as the test blank.
7-3-2 Calculation and interpretation
All data to be included in the data analysis are to relate to cells for which solution G and at least one of solutions A, B and C give a response that is greater than the basal release of the read-out (Solution R0). For each different cell source, e.g. individual donation, donor pool, or cell line, use the standard curve for the read-out (a calibration curve in duplicate with a blank and at least 4 geometrically diluted concentrations of the standard for the chosen read-out) and calculate the mean responses of the replicates of solutions A-F. Sum the mean responses to solutions A, B and C and sum the mean responses to solutions D, E and F. Divide the sum of the mean responses to solutions D, E and F by the sum of the mean responses to solutions A, B and C. The preparation being examined complies with the test for a given cell source if the resulting value complies with a defined acceptance criterion not exceeding a justified value, e.g. 2.5.
7-3-3 Pass/fail criteria of the preparation
The criteria are the same as for method A (see 7-1-3).
To quantify more closely the level of contamination, Methods A, B and C may be performed using other dilutions of the solution of the preparation being examined not exceeding the MVD.
Guidance notes
1 Introduction
The monocyte-activation test (MAT) is primarily intended to be used as a replacement for the rabbit pyrogen test. The MAT detects pyrogenic and pro-inflammatory contaminants, including endotoxins from gram-negative bacteria and ‘non-endotoxin′ contaminants, including pathogen-associated molecular patterns (PAMPs), derived from gram-positive and gram-negative bacteria, viruses and fungi, and product-related and process-related biological or chemical entities.
Since non-endotoxin contaminants are a physico-chemically diverse class of molecules, and usually the nature of the contaminant in a preparation being examined is unknown, the level of contamination is expressed either in endotoxin-equivalent units, derived by comparison with responses to standard endotoxin, or by comparison with a reference lot of the preparation being examined.
In the MAT, responses to standard endotoxin usually dilute out over approximatively 1 log10 and responses to products contaminated with non-endotoxin contaminants (alone or in combination with endotoxins) often show very steep dose-response curves, usually over only 1 or 2 dilution steps when tested for their capability to stimulate monocytes. Frequently, the largest response to such contaminated products is obtained with undiluted solutions of preparations being examined or small dilutions of the preparations being examined. For this reason test solutions of preparations being examined that contain or may contain non-endotoxin contaminants have to be tested at a range of dilutions that includes minimum dilution.
2 Methods
2-1 INFORMATION REGARDING THE choice of methods
Methods A, B and C, are not normally applied where a preparation being examined has the intrinsic activity of stimulating the release of the chosen read-out or where the preparation being examined is contaminated with the chosen read-out. In both cases, this fact is to be addressed by modifying and validating the chosen method accordingly. The product-specific validation of the chosen method would be expected to identify the frequency of non-responders to a particular product/contaminant(s) combination and to identify steps to address this, e.g. screening of donors, increasing the number of donors per test, and setting pass/fail criteria of appropriate stringency to maximise the likelihood of detecting contaminated batches. Method A is not appropriate if the results of different dilutions (endotoxin equivalents per millilitre) show that the dose response curve is not parallel to the standard endotoxin curve. Method B is a semi-quantitative test that can be applied when responses to dilutions of a preparation being examined are not parallel to responses to dilutions of standard endotoxin.
Method C, the reference lot comparison test, was developed to address extreme donor variability in responses to certain product/contaminant(s) combinations. In this regard, it should be noted that, while monocytes from most donors respond in a broadly similar manner to bacterial endotoxin, responses of monocytes from different donors to non-endotoxin contaminants can differ markedly, so that it is possible to identify non-responders along with low and high responders to certain product/contaminant(s) combinations.
2-2 CALCULATION OF CONTAMINANT LIMIT CONCENTRATION
The acceptance criterion for a pass/fail decision is the contaminant limit concentration (CLC), which is expressed in endotoxin equivalents per milligram or millilitre or in units of biological activity of the preparation being examined. Where an endotoxin limit concentration (ELC) has been specified for a product, the CLC is the same as the ELC, unless otherwise prescribed. The CLC is expressed in terms of endotoxin equivalents. The CLC is calculated using the following expression:
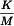
| K | = | threshold pyrogenic dose per kilogram of body mass; |
| M | = | maximum recommended bolus dose of product per kilogram of body mass. |
When the product is to be injected at frequent intervals or infused continuously, M is the maximum total dose administered in a single hour period.
The CLC depends on the product and its route of administration and is stated in some monographs.
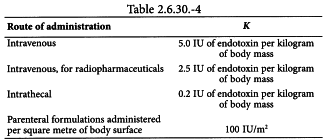
Values for K are suggested in Table 2.6.30.-4.
2-3 Information regarding cryo-protectants
The influence of cryo-protectants, e.g. dimethyl sulfoxide (DMSO), and their residues in thawed cells, is to be considered: DMSO is toxic to cells in culture and, even when cells have been washed thoroughly, cryo-preservation may have altered cell properties, e.g. cell membrane permeability.
2-4 Interference testing
Where practicable, interference testing is performed on at least 3 different lots of the preparation being examined. Preparations being examined that show marked batch-to-batch variation, that effectively renders each batch unique for the purposes of interference testing, are to be subjected to interference testing within each individual test, i.e. concomitant validation.
Interference testing is preferably performed on batches of the preparation being examined that are free of endotoxins and other pyrogenic/pro-inflammatory contaminants and, where this is not practicable, none of the batches are to be heavily contaminated. If only 1 batch is available the validation has to be performed on that batch in 3 independent tests. Precision parameters for reproducibility, e.g. ± 50 per cent, are to be fulfilled.
2-5 Cross-validation
General chapter 5.1.10. Guidelines for using the test for bacterial endotoxins, states that an MAT should be performed on products where the presence of non-endotoxin pyrogenic substances cannot be ruled out. It is therefore recommended to perform cross-validation experiments with the MAT together with the validation experiments for the test for bacterial endotoxins (BET) using the same 3 batches. Cross-validation should be repeated on 3 batches if important process parameters have changed, as potential contamination by non-endotoxin pyrogens cannot be ruled out.
The rabbit pyrogen test (see general chapter 2.6.8. Pyrogens) must only be performed for cross validation if none of the MAT methods (A, B or C) can be validated for a certain product.
3 Replacement of the rabbit pyrogen test by the monocyte activation test
As noted above, MAT is primarily intended to be used as a replacement for the rabbit pyrogen test. Monographs on pharmaceutical products intended for parenteral administration that may contain pyrogenic contaminants require either a test for bacterial endotoxins or a monocyte activation test.
As a general policy:
4 Validation of alternative methods
Replacement of a rabbit pyrogen test, or replacement of a method for detecting pro-inflammatory/pyrogenic contaminants by another method, is to be regarded as the use of an alternative method in the replacement of a pharmacopoeial test, as described in the General Notices.
The following procedures are suggested for validating a method for the MAT other than the one indicated in the monograph:
MAT should be applied to all new products intended for parenteral administration that have to be tested for the presence of non-endotoxin pyrogens according to the requirements of the European Pharmacopoeia.