Appendix XIV J. Blood and Related Products
Coagulants
A1. Assay of human coagulation factor II
Human coagulation factor II is assayed following specific activation to form factor IIa. Factor IIa is estimated by comparing its activity in cleaving a specific chromogenic peptide substrate with the same activity of the International Standard or of a reference preparation calibrated in International Units.
The International Unit is the factor II activity of a stated amount of the International Standard which consists of a freeze-dried concentrate of human blood coagulation factor II. The equivalence in International Units of the International Standard is stated by the World Health Organization.
The chromogenic assay method consists of 2 steps: snake venom-dependent activation of factor II, followed by enzymatic cleavage of a chromogenic factor IIa substrate to form a chromophore that can be quantified spectrophotometrically. Under appropriate assay conditions, there is a linear relation between factor IIa activity and the cleavage of the chromogenic substrate.
REAGENTS
Viper venom specific factor II activator (ecarin) A protein derived from the venom of the saw-scaled viper (Echis carinatus) which specifically activates factor II. Reconstitute according to the manufacturer’s instructions. Store the reconstituted preparation at 4 °C and use within 1 month.
Factor IIa chromogenic substrate Specific chromogenic substrate for factor IIa such as: H-d-phenylalanyl-l-pipecolyl-l-arginine-4-nitroanilide dihydrochloride, 4-toluenesulfonyl-glycyl-prolyl-l-arginine-4-nitroanilide, H-d-cyclohexylglycyl-α-aminobutyryl-l-arginine-4-nitroanilide, d-cyclohexylglycyl-l-alanyl-l-arginine-4-nitroanilide diacetate. Reconstitute according to the manufacturer’s instructions.
Dilution buffer Solution containing 6.06 g/L of tris(hydroxymethyl)aminomethane R, 17.53 g/L of sodium chloride R and 1 g/L of bovine albumin R or human albumin R. Adjust to pH 8.4 if necessary, using hydrochloric acid R.
METHOD
Test solution Dilute the preparation to be examined with dilution buffer to obtain a solution containing 0.015 IU of factor II per millilitre. Prepare at least 3 further dilutions in dilution buffer.
Reference solution Dilute the reference preparation to be examined with dilution buffer to obtain a solution containing 0.015 IU of factor II per millilitre. Prepare at least 3 further dilutions in dilution buffer.
Warm all solutions to 37 °C in a water-bath shortly before the test.
The following working conditions apply to microtitre plates. If the assay is carried out in tubes, the volumes are adjusted while maintaining the proportions in the mixture.
Using a microtitre plate maintained at 37 °C, add 25 µL of each dilution of the test solution or the reference solution to each of a series of wells. To each well add 125 µL of dilution buffer, then 25 µL of ecarin and incubate for exactly 2 min. To each well add 25 µL of factor IIa chromogenic substrate.
Read the rate of change of absorbance (2.2.25) at 405 nm continuously over a period of 3 min and obtain the mean rate of change of absorbance (ΔA/min). If continuous monitoring is not possible, read the absorbance at 405 nm at suitable consecutive intervals, for instance 40 s, plot the absorbances against time on a linear graph and calculate ΔA/min as the slope of the line. From the ΔA/min values of each individual dilution of standard and test preparations, calculate the potency of the preparation to be examined and check the validity of the assay by the usual statistical methods (5.3).
A2. Assay of human coagulation factor VII
Human coagulation factor VII is assayed by its biological activity as a factor VIIa-tissue factor complex in the activation of factor X in the presence of calcium ions and phospholipids. The potency of a factor VII preparation is estimated by comparing the quantity necessary to achieve a certain rate of factor Xa formation in a test mixture containing the substances that take part in the activation of factor X, and the quantity of the International Standard, or of a reference preparation calibrated in International Units, required to produce the same rate of factor Xa formation.
The International Unit is the factor VII activity of a stated amount of the International Standard, which consists of freeze-dried plasma. The equivalence in International Units of the International Standard is stated by the World Health Organization.
Human coagulation factor VII concentrate BRP is calibrated in International Units by comparison with the International Standard.
The chromogenic assay method consists of 2 consecutive steps: the factor VII-dependent activation of factor X reagent mixture containing tissue factor, phospholipids and calcium ions, followed by enzymatic cleavage of a chromogenic factor Xa substrate into a chromophore that can be quantified spectrophotometrically. Under appropriate assay conditions, there is a linear relation between the rate of factor Xa formation and the factor VII concentration. The assay is summarised in the following scheme.
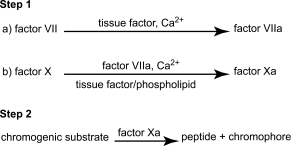
Both steps employ reagents that may be obtained commercially from a variety of sources. Although the composition of individual reagents may be subject to some variation, their essential features are described in the following specification.
REAGENTS
The coagulation factor reagent comprises purified proteins derived from human or bovine sources. These include factor X and thromboplastin tissue factor/phospholipid as factor VII activator. These proteins are partly purified and do not contain impurities that interfere with the activation of factor VII or factor X. Factor X is present in amounts giving a final concentration during the first step of the assay of 10-350 nmol/L, preferably 14-70 nmol/L. Thromboplastin from natural sources (bovine or rabbit brain) or synthetic preparations may be used as the tissue factor/phospholipid component. Thromboplastin suitable for use in prothrombin time determination is diluted 1:5 to 1:50 in buffer such that the final concentration of Ca2+ is 15-25 mmol/L. The final factor Xa generation is performed in a solution containing human or bovine albumin at a concentration such that adsorption losses do not occur and which is appropriately buffered at pH 7.3-8.0. In the final incubation mixture, factor VII must be the only rate-limiting component and each reagent component must lack the ability to generate factor Xa on its own.
The second step comprises the quantification of the formed factor Xa employing a chromogenic substrate that is specific for factor Xa. Generally this consists of a short peptide of between three and five amino acids, bound to a chromophore group. On cleavage of this group from the peptide substrate, its absorption maximum shifts to a wavelength allowing its spectrophotometric quantification. The substrate is usually dissolved in water R and used at a final concentration of 0.2-2 mmol/L. The substrate may also contain appropriate inhibitors to stop further factor Xa generation (addition of edetate).
ASSAY PROCEDURE
Reconstitute the entire contents of one ampoule of the reference preparation and the preparation to be examined by adding the appropriate quantity of water R; use within 1 h. Add sufficient prediluent to the reconstituted preparations to produce solutions containing between 0.5 IU and 2.0 IU of factor VII per millilitre.
Prepare further dilutions of reference and test preparations using an isotonic non-chelating buffer containing 1 per cent of bovine or human albumin, buffered preferably between pH 7.3 and 8.0. Prepare at least three separate, independent dilutions for each material, preferably in duplicate. Prepare the dilutions such that the final factor VII concentration is below 0.005 IU/mL.
Prepare a control solution that includes all components except factor VII.
Prepare all dilutions in plastic tubes and use within 1 h.
Step 1
Mix dilutions of the factor VII reference preparation and the preparation to be examined with an appropriate volume of the prewarmed coagulation factor reagent or a combination of its separate constituents, and incubate the mixture in plastic tubes or microplate wells at 37 °C. The concentrations of the various components during the factor Xa generation must be as specified above under the description of the reagents.
Allow the activation of factor X to proceed for a suitable time, usually terminating the reaction before the factor Xa concentration has reached its maximal level in order to obtain a satisfactory linear dose-response relationship. The activation time is also chosen to achieve linear production of factor Xa in time. Appropriate activation times are usually between 2 min and 5 min, but deviations are permissible if acceptable linearity of the dose-response relationship is thus obtained.
Step 2
Terminate the activation by the addition of a prewarmed reagent containing a chromogenic substrate. Quantify the rate of substrate cleavage, which must be linear with the concentration of factor Xa formed, by measuring the absorbance change at an appropriate wavelength using a spectrophotometer, either monitoring the absorbance continuously, thus allowing the initial rate of substrate cleavage to be calculated, or terminating the hydrolysis reaction after a suitable interval by lowering the pH by the addition of a suitable reagent, such as acetic acid (500 g/L C2H4O2) or a citrate solution (1 mol/L) at pH 3. Adjust the hydrolysis time to achieve a linear development of chromophore with time. Appropriate hydrolysis times are usually between 3 min and 15 min, but deviations are permissible if better linearity of the dose-response relationship is thus obtained.
Check the validity of the assay and calculate the potency of the test preparation by the usual statistical methods (for example, 5.3).
A3. Assay of factor VIII fraction (human coagulation factor VIII)
Human coagulation factor VIII is assayed by its biological activity as a cofactor in the activation of factor X by activated factor IX (factor IXa) in the presence of calcium ions and phospholipid. Factor VIII activity may be measured in plasma preparations and therapeutic concentrates (plasma-derived and recombinant). The potency of a factor VIII preparation is estimated by comparing the quantity necessary to achieve a certain rate of factor Xa formation in a test mixture containing the substances that take part in the activation of factor X, and the quantity of the International Standard, or of a reference preparation calibrated in International Units, required to produce the same rate of factor Xa formation.
Quantification of factor VIII activity in plasma preparations is expressed in International Units defined by the International Standard for blood coagulation factor VIII in plasma, and coagulation factors V, VIII, XI and XIII plasma BRP is suitable for use as a reference preparation. Quantification of factor VIII activity in therapeutic concentrates is expressed in International Units defined by the International Standard for blood coagulation factor VIII concentrate, and human coagulation factor VIII concentrate BRP is suitable for use as a reference preparation.
The chromogenic assay method consists of 2 consecutive steps: the factor VIII-dependent activation of factor X in a coagulation-factor reagent composed of purified components, and the enzymatic cleavage of a chromogenic factor Xa substrate to yield a chromophore that can be quantified spectrophotometrically. Under appropriate assay conditions, there is a linear relation between the rate of factor Xa formation and the factor VIII concentration. The assay is summarised by the following scheme.
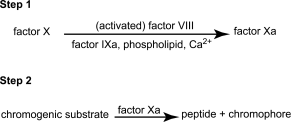
Both steps employ reagents that may be obtained commercially from a variety of sources. Although the composition of individual reagents may be subject to some variation, their essential features are described in the following specification. Deviations from this description may be permissible provided that it has been shown, using the appropriate International Standard for blood coagulation factor VIII as the standard, that the results obtained do not differ significantly.
It is important to demonstrate by validation the suitability of the kit used, notably by checking the time course of factor Xa generation in order to determine the time taken to reach 50 per cent of the maximal factor Xa generation.
REAGENTS
The coagulation factor reagent comprises purified proteins derived from human or bovine sources. These include factor X, factor IXa, and a factor VIII activator, usually thrombin. These proteins are partly purified, preferably to at least 50 per cent, and do not contain impurities that interfere with the activation of factor VIII or factor X. Thrombin may be present in its precursor form prothrombin, provided that its activation in the reagent is sufficiently rapid to give almost instantaneous activation of factor VIII in the assay. Phospholipid may be obtained from natural sources or be synthetically prepared, and must, to a substantial extent, consist of the species phosphatidylserine. The components of the complete reagent are usually divided into at least 2 separate reagents, each lacking the ability to generate factor Xa on its own. One of the reagents contains calcium ions. After reconstitution, the reagents may be combined provided that no substantial amounts of factor Xa are generated in the absence of factor VIII. In the final incubation mixture, factor VIII must be the only rate-limiting component.
The 2nd step comprises the quantification of the formed factor Xa, employing a chromogenic substrate that is specific for factor Xa. Generally this consists of a derivatised short peptide of between 3 and 5 amino acids, joined to a chromophore group. On cleavage of this group from the peptide substrate, its chromophoric properties shift to a wavelength allowing its spectrophotometric quantification. The substrate must also contain appropriate inhibitors to stop further factor Xa generation, e.g. chelating agents, and to suppress thrombin activity.
ASSAY PROCEDURE
For the assay of therapeutic concentrates, add sufficient prediluent to the reference and test preparations to produce solutions containing 0.5-2.0 IU/mL. The prediluent consists of haemophilia A plasma, or of an artificially prepared reagent that contains sufficient von Willebrand factor and that gives results that do not differ significantly from those obtained employing haemophilia plasma. The prediluted materials must be stable beyond the time required for the assay. Pre-dilution in haemophilia A plasma is not required for the assay of factor VIII in plasma preparations.
Prepare dilutions of the pre-diluted reference and test concentrate preparations (or the reference and test plasma preparations) using a non-chelating, appropriately buffered solution, for example, tris(hydroxymethyl)aminomethane or imidazole, containing 1 per cent of human or bovine albumin. Prepare at least 2 dilution series of at least 3 further dilutions for each material. Prepare the dilutions such that the final factor VIII concentration in the reaction mixture is preferably below 0.01 IU/mL, during the step of factor Xa generation.
Prepare a control solution that includes all components except factor VIII.
Prepare all dilutions in plastic tubes and use immediately.
Step 1
Mix prewarmed dilutions of the factor VIII reference preparation and of the preparation to be examined with an appropriate volume of the prewarmed coagulation factor reagent or a combination of its separate constituents, and incubate the mixture in plastic tubes or microplate wells at 37 °C. Allow the activation of factor X to proceed for a suitable time, terminating the reaction (step 2) when the factor Xa concentration has reached approximately 50 per cent of the maximal (plateau) level. Appropriate activation times are usually between 2 min and 5 min.
Step 2
Terminate the activation by addition of a prewarmed reagent containing a chromogenic substrate. Quantify the rate of substrate cleavage, which must be linear with the concentration of factor Xa formed, by measuring the absorbance change at an appropriate wavelength using a spectrophotometer, either monitoring the absorbance continuously, thus allowing the initial rate of substrate cleavage to be calculated, or terminating the hydrolysis reaction after a suitable interval by lowering the pH by addition of a suitable reagent, such as a 50 per cent V/V solution of acetic acid, or a 1 M pH 3 citrate buffer solution. Adjust the hydrolysis time to achieve a linear development of chromophore over time. Appropriate hydrolysis times are usually between 3 min and 15 min, but deviations are permissible if better linearity of the dose-response relationship is thus obtained.
Calculate the potency of the test preparation by the usual statistical methods (for example, 5.3).
A4. Assay of factor IX fraction (human coagulation factor IX)
The principle of the assay is to measure the ability of a factor IX preparation to reduce the prolonged coagulation time of factor IX-deficient plasma. The reaction is accelerated by addition of a reagent containing phospholipid and a contact activator, e.g. kaolin, silica or ellagic acid. The potency is assessed by comparing the dose-response curve of the preparation to be examined to that of a reference preparation, calibrated in International Units.
The International Unit is the factor IX activity of a stated amount of the International Standard, which consists of a freeze-dried concentrate of human coagulation factor IX. The equivalence in International Units of the International Standard is stated by the World Health Organization.
Human coagulation factor IX concentrate BRP is calibrated in International Units by comparison with the International Standard.
Reconstitute separately the preparation to be examined and the reference preparation as stated on the label and use immediately. Where applicable, determine the amount of heparin present (2.7.12) and neutralise the heparin, for example by addition of protamine sulfate R (10 µg of protamine sulfate neutralises 1 IU of heparin). Predilute the preparation to be examined and the reference preparation in factor IX-deficient plasma (for example plasma substrate R2) to produce solutions containing 0.5-2.0 IU/mL. Prepare at least 3 dilutions for each material, preferably in duplicate, using a suitable buffer solution (for example imidazole buffer solution pH 7.3 R) containing 10 g/L of bovine or human albumin. Use these dilutions immediately.
Use an apparatus suitable for measurement of coagulation times or carry out the assay with incubation tubes maintained in a water-bath at 37 °C. Place in each tube 0.1 mL of factor IX-deficient plasma (for example plasma substrate R2) and 0.1 mL of one of the dilutions of the reference preparation or of the preparation to be examined. Add to each tube 0.1 mL of a suitable Activated Partial Thromboplastin Time (APTT) reagent containing phospholipid and contact activator and incubate the mixture for a recommended time at 37 °C. To each tube, add 0.1 mL of a 3.7 g/L solution of calcium chloride R previously heated to 37 °C. Using a timer, measure the coagulation time, i.e. the interval between the moment of the addition of the calcium chloride and the first indication of the formation of fibrin. The volumes given above may be adapted to the APTT reagent and apparatus used. Calculate the potency using the usual statistical methods (for example, 5.3).
A5. Assay of human coagulation factor X
Human coagulation factor X is assayed following specific activation to form factor Xa. Factor Xa is estimated by comparing its activity in cleaving a specific chromogenic peptide substrate with the same activity of the International Standard or of a reference preparation calibrated in International Units.
The International Unit is the factor X activity of a stated amount of the International Standard which consists of a freeze-dried concentrate of human coagulation factor X. The equivalence in International Units of the International Standard is stated by the World Health Organization.
The chromogenic assay method consists of 2 steps: snake venom-dependent activation of factor X, followed by enzymatic cleavage of a chromogenic factor Xa substrate to form a chromophore that can be quantified spectrophotometrically. Under appropriate assay conditions, there is a linear relation between factor Xa activity and the cleavage of the chromogenic substrate.
REAGENTS
Russell’s viper venom specific factor X activator (RVV) A protein derived from the venom of Russell’s viper (Vipera russelli) which specifically activates factor X. Reconstitute according to the manufacturer’s instructions. Store the reconstituted preparation at 4 °C and use within 1 month.
Factor Xa chromogenic substrate Specific chromogenic substrate for factor Xa such as: N-α-benzyloxycarbonyl-d-arginyl-l-glycyl-l-arginine-4-nitroanilide dihydrochloride, N-benzoyl-l-isoleucyl-l-glutamyl-glycyl-l-arginine-4-nitroanilide hydrochloride, methanesulfonyl-d-leucyl-glycyl-l-arginine-4-nitroanilide, methoxycarbonyl-d-cyclohexylalanyl-glycyl-l-arginine-4-nitroanilide acetate. Reconstitute according to the manufacturer’s instructions.
Dilution buffer Solution containing 3.7 g/L of tris(hydroxymethyl)aminomethane R, 18.0 g/L of sodium chloride R, 2.1 g/L of imidazole R, 0.02 g/L of hexadimethrine bromide R and 1 g/L of bovine albumin R or human albumin R. Adjust to pH 8.4 if necessary using hydrochloric acid R.
METHOD
Test solution Dilute the preparation to be examined with dilution buffer to obtain a solution containing 0.18 IU of factor X per millilitre. Prepare at least 3 further dilutions in dilution buffer.
Reference solution Dilute the reference preparation to be examined with dilution buffer to obtain a solution containing 0.18 IU of factor X per millilitre. Prepare at least 3 further dilutions in dilution buffer.
Warm all solutions to 37 °C in a water-bath shortly before the test.
The following working conditions apply to microtitre plates. If the assay is carried out in tubes, the volumes are adjusted while maintaining the proportions in the mixture.
Using a microtitre plate maintained at 37 °C, add 12.5 µL of each dilution of the test solution or the reference solution to each of a series of wells. To each well add 25 µL of RVV and incubate for exactly 90 s. To each well add 150 µL of factor Xa chromogenic substrate, diluted 1 in 6 in dilution buffer.
Read the rate of change of absorbance (2.2.25) (at 405 nm continuously over a period of 3 min and obtain the mean rate of change of absorbance (ΔA/min). If continuous monitoring is not possible, read the absorbance at 405 nm at suitable consecutive intervals, for instance 40 s, plot the absorbances against time on a linear graph and calculate ΔA/min as the slope of the line. From the ΔA/min values of each individual dilution of standard and test preparations, calculate the potency of the preparation to be examined and check the validity of the assay by the usual statistical methods (5.3).
A6. Assay of human coagulation factor XI
The principle of the assay is to measure the ability of a factor XI preparation to reduce the prolonged coagulation time of factor XI-deficient plasma. The reaction is accelerated by addition of a reagent containing phospholipid and a contact activator, e.g. kaolin, silica or ellagic acid. The potency is assessed by comparing the dose-response curve of the preparation to be examined to that of a reference plasma calibrated against the International Standard for blood coagulation factor XI in plasma.
Reconstitute separately the preparation to be examined and the reference preparation as stated on the label and use immediately. Coagulation factors V, VIII, XI and XIII plasma BRP is suitable for use as a reference preparation. Where applicable, determine the amount of heparin present (2.7.12) and neutralise the heparin, for example by addition of protamine sulfate R (10 µg of protamine sulfate neutralises 1 IU of heparin). Predilute the preparation to be examined and the reference preparation in factor XI-deficient plasma (for example plasma substrate R3) to produce solutions containing 0.5-2.0 units/mL. Prepare at least 3 appropriate dilutions for each material, preferably in duplicate, using a suitable buffer solution (for example imidazole buffer solution pH 7.3 R) containing 10 g/L of bovine or human albumin. Use these dilutions immediately.
Use an apparatus suitable for measurement of coagulation times or perform the assay with incubation tubes maintained in a water bath at 37 °C. Place in each tube 0.1 mL of factor XI-deficient plasma (for example plasma substrate R3) and 0.1 mL of one of the dilutions of the reference preparation or of the preparation to be examined. Add to each tube 0.1 mL of a suitable Activated Partial Thromboplastin Time (APTT) reagent containing phospholipid and contact activator and incubate the mixture for a recommended time at 37 °C. To each tube, add 0.1 mL of a 3.7 g/L solution of calcium chloride R previously heated to 37 °C. Using a timer, measure the coagulation time, i.e. the interval between the moment of the addition of the calcium chloride and the first indication of the formation of fibrin. The volumes given above may be adapted to the APTT reagent and apparatus used. Calculate the potency using the usual statistical methods (for example, 5.3).
A7. Activated coagulation factors
Where applicable, determine the amount of heparin present (2.7.12) and neutralise the heparin, for example by addition of protamine sulfate R (10 µg of protamine sulfate neutralises 1 IU of heparin). Prepare 1 to 10 and 1 to 100 dilutions of the preparation to be examined using tris(hydroxymethyl)aminomethane buffer solution pH 7.5 R. Place a series of polystyrene tubes in a water-bath at 37 °C and add to each tube 0.1 mL of platelet-poor plasma R and 0.1 mL of a suitable dilution of a phospholipid preparation to act as a platelet substitute. Allow to stand for 60 s. Add to each tube either 0.1 mL of 1 of the dilutions or 0.1 mL of the buffer solution (control tube). To each tube add immediately 0.1 mL of a 3.7 g/L solution of calcium chloride R previously heated to 37 °C, and measure, within 30 min of preparing the original dilution, the time that elapses between addition of the calcium chloride solution and the formation of a clot. The test is not valid unless the coagulation time measured for the control tube is 200 s to 350 s.
A8. Assay of human von Willebrand factor
The biological functions of human von Willebrand factor are numerous. At present, its ristocetin cofactor activity and its collagen binding activity can be utilised for assays. The potency of human von Willebrand factor is determined by comparing, in given conditions, its activity with the same activity of a reference preparation calibrated against the International Standard, in International Units where applicable.
The International Unit is the activity of a stated amount of the International Standard, which consists of a freeze-dried human von Willebrand factor concentrate. The equivalence in International Units of the International Standard is stated by the World Health Organization (WHO).
Ristocetin cofactor assay
The ristocetin cofactor activity of von Willebrand factor is determined by measuring agglutination of a suspension of platelets in the presence of ristocetin A. The assay can be carried out for quantitative determinations by using automated instruments, or for semi-quantitative determinations by visually assessing the endpoint of agglutination in a dilution series. Quantitative assays are preferred.
REAGENTS
Suspension of platelets Use standardised and, for example, formaldehyde- or paraformaldehyde-fixed preparations of freshly isolated and washed human platelets. The suspension may also be freeze-dried. An appropriate amount of ristocetin A is added if necessary. Some platelet reagents may already contain ristocetin A.
Reference preparation The reference preparation for von Willebrand factor is the WHO International Standard for von Willebrand factor concentrate.
METHOD
Semi-quantitative assay Prepare suitable dilutions of the preparation to be examined and of the reference preparation, using as diluent a solution containing 9 g/L of sodium chloride R and 10-50 g/L of human albumin R. Add to each dilution an appropriate amount of the suspension of platelets and, if necessary, of ristocetin A. Mix on a glass slide by moving it gently in circles for 1 min. Allow to stand for a further 1 min and read the result against a dark background with side lighting. The last dilution which clearly shows visible agglutination indicates the ristocetin cofactor titre of the sample. Use diluent as a negative control.
Quantitative Assay Reconstitute the entire contents of 1 ampoule of the reference preparation and the preparation to be examined by adding the appropriate quantity of the recommended diluent (for example water R); use immediately. Add sufficient prediluent to the reconstituted preparations to produce solutions containing 0.5-2.0 IU/mL. The prediluent consists of an isotonic non-chelating buffer containing, for example, 1-5 per cent of human or bovine albumin, and tris(hydroxymethyl)aminomethane or imidazole, appropriately buffered.
The test is performed in accordance with the manufacturer′s instructions with at least 2 dilution series with as many dilutions as are needed to obtain a total of at least 3 different concentrations in the linear range of the assay.
Check the validity of the assay and calculate the potency of the test preparation using the usual statistical methods (for example, 5.3).
Collagen-binding assay
Collagen-binding is determined by an enzyme-linked immunosorbent assay on collagen-coated microtitre plates. The method is based on the specific binding of von Willebrand factor to collagen fibrils and the subsequent binding of polyclonal anti-von Willebrand factor antibody conjugated to an enzyme, which on addition of a chromogenic substrate yields a product that can be quantitated spectrophotometrically. Under appropriate conditions, there is a linear relationship between von Willebrand factor collagen-binding and absorbance.
REAGENTS
Collagen Use native equine or human fibrils of collagen type I or III. For ease of handling, collagen solutions may be used.
Collagen diluent Dissolve 50 g of glucose R in water R, adjust to pH 2.7-2.9 with 1 M hydrochloric acid and dilute to 1000 mL with water R.
Phosphate-buffered saline (PBS) Dissolve 8.0 g of sodium chloride R, 1.05 g of disodium hydrogen phosphate dihydrate R, 0.2 g of sodium dihydrogen phosphate R and 0.2 g of potassium chloride R in water R. Adjust to pH 7.2 using 1 M sodium hydroxide or 1 M hydrochloric acid and dilute to 1000 mL with water R.
Washing buffer PBS containing 1 g/L of polysorbate 20 R.
Blocking reagent PBS containing 1 g/L of polysorbate 20 R and 10 g/L of bovine albumin R.
Dilution buffer PBS containing 1 g/L of polysorbate 20 R and 50 g/L of bovine albumin R.
Conjugate Rabbit anti-human von Willebrand factor serum horseradish peroxidase conjugate. Use according to the manufacturer′s instructions.
Substrate solution Immediately before use, dissolve a tablet of o-phenylenediamine dihydrochloride and a tablet of urea hydrogen peroxide in 20 mL of water R or use a suitable volume of hydrogen peroxide. Protect from light.
Microtitre plates Flat-bottomed polystyrene plates with surface properties optimised for enzyme immunoassay and high protein-binding capacity.
METHOD
Test solutions Reconstitute the preparation to be examined as stated on the label. Dilute with dilution buffer to produce a solution containing approximately 1 IU of von Willebrand factor. Prepare 2 series of at least 3 further dilutions using dilution buffer.
Reference solutions Reconstitute the reference preparation as directed. Dilute with dilution buffer to produce a solution containing approximately 1 IU of von Willebrand factor. Prepare 2 series of at least 3 further dilutions using dilution buffer.
Allow the solution of collagen to warm to room temperature. Dilute with collagen diluent to obtain a solution containing 30-75 µg/mL of collagen, mix gently to produce a uniform suspension of collagen fibrils. Pipette 100 µL into each well of the microtitre plate. Cover the plate with plastic film and incubate at 37 °C overnight. Empty the wells of the collagen-coated plate by inverting and draining on a paper towel. Add 250 µL of washing buffer. Empty the wells of the plate by inverting and draining on a paper towel. Repeat this operation 3 times. Add 250 µL of blocking reagent to each well, cover the plate with plastic film and incubate at 37 °C for 1 h. Empty the wells of the plate by inverting and draining on a paper towel. Add 250 µL of washing buffer. Empty the wells of the plate by inverting and draining on a paper towel. Repeat this operation 3 times.
Add 100 µL each of the test solutions or reference solutions to the wells. Add 100 µL of dilution buffer to a series of wells to serve as negative control. Cover the plate with plastic film and incubate at 37 °C for 2 h. Empty the wells of the plate by inverting and draining on a paper towel. Add 250 µL of washing buffer. Empty the wells of the plate by inverting and draining on a paper towel. Repeat this operation 3 times.
Prepare a suitable dilution of the conjugate (for example, a dilution factor of 1 to 4000) with PBS containing 5 g/L of bovine albumin R and add 100 µL to each well. Cover the plate with plastic film and incubate at 37 °C for 2 h. Empty the wells of the plate by inverting and draining on a paper towel. Add 250 µL of washing buffer. Empty the wells of the plate by inverting and draining on a paper towel. Repeat this operation 3 times.
Add 100 µL of substrate solution to each of the wells and incubate at room temperature for 20 min in the dark. Add 100 µL of 1 M hydrochloric acid to each of the wells.
Measure the absorbance at 492 nm. Use the absorbance values to estimate the potency of the preparation to be examined using the usual statistical methods (5.3).
The assay is invalid if the absorbances measured for the negative controls are greater than 0.05.
A9. Test for prekallikrein activator
Prekallikrein activator (PKA) activates prekallikrein to kallikrein and may be assayed by its ability to cleave a chromophore from a synthetic peptide substrate so that the rate of cleavage can be measured spectrophotometrically and the concentration of PKA calculated by comparison with a reference preparation calibrated in International Units.
The International Unit is the activity of a stated amount of the International Standard, which consists of freeze-dried prekallikrein activator. The equivalence in International Units of the International Standard is stated by the World Health Organization.
REAGENTS
Prekallikrein activator in albumin BRP is calibrated in International Units by comparison with the International Standard.
Buffer A Dissolve 6.055 g of tris(hydroxymethyl)aminomethane R, 1.17 g of sodium chloride R, 50 mg of hexadimethrine bromide R and 0.100 g of sodium azide R in water R. Adjust to pH 8.0 with 2 M hydrochloric acid R and dilute to 1000 mL with water R.
Buffer B Dissolve 6.055 g of tris(hydroxymethyl)aminomethane R and 8.77 g of sodium chloride R in water R. Adjust to pH 8.0 with 2 M hydrochloric acid R and dilute to 1000 mL with water R.
PREPARATION OF PREKALLIKREIN SUBSTRATE
To avoid coagulation activation, blood or plasma used for the preparation of prekallikrein must come into contact only with plastics or silicone-treated glass surfaces.
Draw 9 volumes of human blood into 1 volume of anticoagulant solution (ACD, CPD or a 38 g/L solution of sodium citrate R) to which 1 mg/mL of hexadimethrine bromide R has been added. Centrifuge the mixture at 3600 g for 5 min. Separate the plasma and centrifuge again at 6000 g for 20 min to sediment platelets. Separate the platelet-poor plasma and dialyse against 10 volumes of buffer A for 20 h. Apply the dialysed plasma to a chromatography column containing agarose-DEAE for ion-exchange chromatography R that has been equilibrated in buffer A and is equal to twice the volume of the plasma. Elute from the column with buffer A at 20 mL/cm2/h. Collect the eluate in fractions and record the absorbance at 280 nm (2.2.25). Pool the fractions containing the 1st protein peak so that the volume of the pool is about 1.2 times the volume of the platelet-poor plasma.
Test the substrate pool for absence of kallikrein activity by mixing 1 part with 20 parts of the pre-warmed chromogenic substrate solution to be used in the assay and incubate at 37 °C for 2 min. The substrate is suitable if the increase in absorbance is less than 0.001 per minute. Add to the pooled solution 7 g/L of sodium chloride R and filter through a membrane filter (nominal pore size 0.45 µm). Freeze the filtrate in portions and store at -25 °C; the substrate may be freeze-dried before storage.
Carry out all procedures from the beginning of the chromatography to freezing in portions during a single working day.
METHOD
The assay may be carried out using an automated enzyme analyser or a suitable microtitre plate system allowing kinetic measurements, with appropriate software for calculation of results. Standards, samples and prekallikrein substrate may be diluted as necessary using buffer B.
Incubate diluted standards or samples with prekallikrein substrate for 10 min such that the volume of the undiluted sample does not exceed 1/10 of the total volume of the incubation mixture to avoid errors caused by variation in ionic strength and pH in the incubation mixture. Incubate the mixture or a part thereof with at least an equal volume of a solution of a suitable synthetic chromogenic substrate, known to be specific for kallikrein (for example, N-benzoyl-l-prolyl-l-phenylalanyl-l-arginine 4-nitroanilide acetate R or d-prolyl-l-phenylalanyl-l-arginine 4-nitroanilide dihydrochloride R), dissolved in buffer B. Record the rate of change in absorbance per minute for 2-10 min at the wavelength specific for the substrate used. Prepare a blank for each mixture of sample or standard using buffer B instead of prekallikrein substrate.
Depending on the method used, ΔA/min has to be corrected by subtracting the value obtained for the corresponding blank without the prekallikrein substrate. The results may be calculated using a standard curve, a parallel-line or a slope ratio assay or any other suitable statistical method. Plot a calibration curve using the values thus obtained for the reference preparation and the respective concentrations; use the curve to determine the PKA activity of the preparation to be examined. During method validation, the spiking experiments must show that the sample matrix has no influence on the results. High blank values may impact assay validity and should be appropriately investigated.
A10. Assay of human plasmin inhibitor
Human plasmin inhibitor, also called human α2-antiplasmin, is a plasma protein that inhibits the plasmin (a serine protease) pathway of fibrinolysis by rapidly forming a complex with free plasmin. Furthermore, upon blood coagulation, human plasmin inhibitor is cross-linked to fibrin strands by factor XIII, and interferes with binding of the proenzyme plasminogen to fibrin.
The potency of human plasmin inhibitor is estimated by comparing the ability of the preparation to be examined to inhibit the cleavage of a specific chromogenic substrate by plasmin with the same ability of a reference standard of human plasmin inhibitor. Plasmin cleavage of the chromogenic substrate yields a chromophore that can be quantified spectrophotometrically.
The individual reagents for the assay may be obtained separately or in commercial kits. Both end-point and kinetic methods are available. Procedures and reagents may vary between different kits and the manufacturer’s instructions are followed. The essential features of the procedure are described in the following example of a microtitre-plate kinetic method.
reagents
Dilution buffer pH 7.5 According to the manufacturer’s instructions, a suitable buffer is used. Adjust the pH if necessary.
Plasmin A preparation of human plasmin that does not contain significant amounts of other proteases is preferably used. Reconstitute and store according to the manufacturer’s instructions.
Plasmin chromogenic substrate A suitable specific chromogenic substrate for plasmin is used: H-D-cyclohexylalanyl-norvalyl-lysyl-p-nitroaniline hydrochloride (H-D-CHA-Nva-Lys-pNA.HCl) or L-pyroglutamyl-L-phenylalanyl-L-lysyl-p-nitroaniline hydrochloride (Glp-Phe-Lys-pNA.HCl). Reconstitute in water R to give a suitable concentration according to the manufacturer’s instructions.
Method
Varying quantities of the preparation to be examined are mixed with a given quantity of plasmin and the remaining plasmin activity is determined using a suitable chromogenic substrate.
Reconstitute or thaw the preparation to be examined according to the manufacturer′s instructions. Dilute with dilution buffer pH 7.5 and prepare at least 2 independent series of 3 or 4 dilutions for both the preparation to be examined and the reference standard.
Mix 0.020 mL of each dilution with 0.020 mL of dilution buffer pH 7.5 and warm to 37 °C. Add 0.040 mL of a plasmin solution (test concentration in the range of 0.2 nkat/mL to 1.6 nkat/mL) previously heated to 37 °C and leave at 37 °C for 1 min. Add 0.020 mL of the chromogenic substrate solution, previously heated to 37 °C, to each mixture. Immediately start measurement of the change in absorbance at 405 nm (2.2.25) using a microtitre plate reader. Calculate the rate of change of absorbance (ΔA/min). Alternatively, an end-point assay might be used by stopping the reaction with acetic acid and measuring the absorbance at 405 nm.
In both cases the duration of the cleavage of the chromogenic substrate should be chosen to produce a linear increase in absorbance at 405 nm, before substrate depletion becomes significant. If the assay is performed in test tubes or cuvettes using a spectrophotometric method, the volumes of reagent solutions are changed proportionally.
Substract the optical density of the blank (prepared with dilution buffer pH 7.5) from the optical density of the preparation to be examined. Check the validity of the assay and calculate the potency of the preparation to be examined by the usual statistical methods (5.3).
Anticoagulants
B1. Assay of heparin in coagulation factors
Heparin is assayed as a complex with antithrombin III (AT) via its inhibition of coagulation factor Xa (anti-Xa activity). An excess of AT is maintained in the reaction mixture to ensure a constant concentration of the heparin-AT complex. Factor Xa is neutralised by the heparin-AT complex and the residual factor Xa hydrolyses a specific chromogenic peptide substrate to release a chromophore. The quantity of chromophore is inversely proportional to the activity of the heparin.
Factor Xa chromogenic substrate Specific chromogenic substrate for factor Xa such as: N-benzoyl-l-isoleucyl-l-glutamyl-glycyl-l-arginine-4-nitroanilide hydrochloride. Reconstitute according to the manufacturer’s instructions.
Dilution buffer 6.05 g/L solution of tris(hydroxymethyl)aminomethane R. Adjust to pH 8.4 if necessary using hydrochloric acid R.
Test solution Dilute the preparation to be examined with dilution buffer to obtain a solution expected to contain 0.1 IU of heparin per millilitre.
Reference solution Dilute the heparin reference preparation with dilution buffer to obtain a solution containing 0.1 IU of heparin per millilitre.
The following working conditions apply to microtitre plates. If the assay is carried out in tubes, the volumes are adjusted while maintaining the proportions in the mixture.
Warm all solutions to 37 °C in a water-bath shortly before the test.
Distribute in a series of wells, 20 µL of normal human plasma and 20 µL of antithrombin III solution R1. Add to the wells a series of volumes (20 µL, 60 µL, 100 µL and 140 µL) of the test solution or the reference solution and make up the volume in each well to 200 µL using dilution buffer (0.02-0.08 IU of heparin per millilitre in the final reaction mixture).
End-point method Transfer 40 µL from each well to a second series of wells, add 20 µL of bovine factor Xa solution R and incubate at 37 °C for 30 s. Add 40 µL of a 1 mmol/L solution of factor Xa chromogenic substrate and incubate at 37 °C for 3 min. Terminate the reaction by lowering the pH by the addition of a suitable reagent, such as a 20 per cent V/V solution of glacial acetic acid R and measure the absorbance at 405 nm (2.2.25). Appropriate reaction times are usually between 3 min and 15 min, but deviations are permissible if better linearity of the dose-response relationship is thus obtained.
Kinetic method Transfer 40 µL from each well to a second series of wells, add 20 µL of bovine factor Xa solution R and incubate at 37 °C for 30 s. Add 40 µL of a 2 mmol/L solution of factor Xa chromogenic substrate, incubate at 37 °C and measure the rate of substrate cleavage by continuous measurement of the absorbance change at 405 nm (2.2.25), thus allowing the initial rate of substrate cleavage to be calculated. This rate must be linear with the concentration of residual factor Xa.
Check the validity of the assay and calculate the heparin activity of the test preparation by the usual statistical methods for a slope-ratio assay (for example, 5.3).
B2. Assay of heparin
The anticoagulant activity of heparin is determined in vitro by its ability to accelerate the inhibition of thrombin, factor IIa (anti-IIa assay), by antithrombin. The International Unit is the activity contained in a stated amount of the International Standard for unfractionated heparin. Heparin sodium BRP, calibrated in International Units by comparison with the International Standard using the 2 assays given below, is used as the reference preparation.
The assay of anti-factor Xa activity is carried out to determine the ratio of anti-factor Xa activity to anti-factor IIa activity.
For anti-IIa and anti-Xa assays, carry out the assay by determining the absorbance (end-point method) or the change of absorbance per minute (kinetic method).
ANTI-FACTOR IIa ACTIVity
Reference and test solutions
Prepare 4 independent series of 4 dilutions each of the substance to be examined and of heparin sodium BRP in tris(hydroxymethyl)aminomethane-EDTA buffer solution pH 8.4 R1; a concentration range within 0.005 IU and 0.03 IU per millilitre is suitable. The dilutions chosen must give a linear response when results are plotted as absorbance against log concentration.
Procedure
Label 16 tubes for the dilutions of the substance to be examined and 16 tubes for the dilutions of the reference preparation: T1, T2, T3, T4 for each of the 4 series of dilutions of the substance to be examined and S1, S2, S3, S4 for each of the 4 series of dilutions of the reference preparation. To each of the 32 tubes add 100 µL of antithrombin III solution R5 and 50 µL of the appropriate dilution of the substance to be examined or the reference preparation. After each addition, mix but do not allow bubbles to form. Treating the tubes in 2 subsequent series in the order S1, S2, S3, S4, T1, T2, T3, T4, T1, T2, T3, T4, S1, S2, S3, S4, allow to equilibrate at 37 °C (water-bath or heating block) for at least 1 min and add to each tube 25 µL of human thrombin solution R2. Incubate for exactly 1 min and add 50 µL of a chromogenic substrate specific to factor IIa at a concentration suitable for the assay (for example, D-phenylalanyl-L-pipecolyl-L-arginine-4-nitroanilide dihydrochloride dissolved in water R to give a 1.25 mM solution).
For the kinetic method, transfer the mixtures to semi-micro cuvettes and measure the change in absorbance per minute (2.2.25) at 405 nm using a suitable reading device.
For the end-point method, stop the reaction after exactly 4 min by adding 50 µL of a 20 per cent V/V solution of glacial acetic acid R. Assess whether exactly 4 min of incubation with the chromogenic substrate yields the optimal absorbance reading and, if necessary, adjust the incubation time to give the best dose-response curve. Then, transfer the mixtures to semi-micro cuvettes and measure the absorbance (2.2.25) at 405 nm using a suitable reading device.
Determine the blank amidolytic activity at the beginning and at the end of the procedure in a similar manner, using tris(hydroxymethyl)aminomethane-EDTA buffer solution pH 8.4 R1 instead of the reference and test solutions; the 2 blank values do not differ significantly.
Calculate the regression of the absorbance on log concentrations of the solutions of the substance to be examined and of heparin sodium BRP, and calculate the potency of the substance to be examined in International Units per millilitre using the usual statistical methods for parallel-line assays (5.3).
ANTI-FACTOR xa ACTIVity
Reference and test solutions
Prepare 4 independent series of 4 dilutions each of the substance to be examined and of heparin sodium BRP in tris(hydroxymethyl)aminomethane-EDTA buffer solution pH 8.4 R1; a concentration range within 0.03 IU and 0.375 IU per millilitre is suitable. The dilutions chosen must give a linear response when results are plotted as absorbance against log concentration.
Procedure
Label 16 tubes for the dilutions of the substance to be examined and 16 tubes for the dilutions of the reference preparation: T1, T2, T3, T4 for each of the 4 series of dilutions of the substance to be examined and S1, S2, S3, S4 for each of the 4 series of dilutions of the reference preparation. To each of the 32 tubes add 50 µL of antithrombin III solution R6 and 50 µL of the appropriate dilution of the substance to be examined or the reference preparation. After each addition, mix but do not allow bubbles to form. Treating the tubes in 2 subsequent series in the order S1, S2, S3, S4, T1, T2, T3, T4, T1, T2, T3, T4, S1, S2, S3, S4, allow to equilibrate at 37 °C (water-bath or heating block) for 1 min and add to each tube 100 µL of bovine factor Xa solution R2. Incubate for exactly 2 min and add 100 µL of a chromogenic substrate specific to factor Xa at a concentration suitable for the assay (for example, N-α-benzyloxycarbonyl-D-arginyl-L-glycyl-L-arginine-4-nitroanilide dihydrochloride dissolved in water R to give a 1 mM solution).
For the kinetic method, transfer the mixtures to semi-micro cuvettes and measure the change in absorbance per minute (2.2.25) at 405 nm using a suitable reading device.
For the end-point method, stop the reaction after exactly 4 min by adding 50 µL of a 20 per cent V/V solution of glacial acetic acid R. Assess whether exactly 4 min of incubation with the chromogenic substrate yields the optimal absorbance reading and, if necessary, adjust the incubation time to give the best dose-response curve. Then, transfer the mixtures to semi-micro cuvettes and measure the absorbance (2.2.25) at 405 nm using a suitable reading device.
Determine the blank amidolytic activity at the beginning and at the end of the procedure in a similar manner, using tris(hydroxymethyl)aminomethane-EDTA buffer solution pH 8.4 R1 instead of the reference and test solutions; the 2 blank values do not differ significantly.
Calculate the regression of the absorbance on log concentrations of the solutions of the substance to be examined and of heparin sodium BRP, and calculate the potency of the substance to be examined in International Units per millilitre using the usual statistical methods for parallel-line assays (5.3).
B3. Assay of human antithrombin III
The antithrombin III content of the preparation to be examined is determined by comparing its ability to inactivate thrombin in the presence of an excess of heparin with the same ability of a reference preparation of human antithrombin III concentrate calibrated in International Units. Varying quantities of the preparation to be examined are mixed with a given quantity of thrombin and the remaining thrombin activity is determined using a suitable chromogenic substrate.
The International Unit is the activity of a stated amount of the International Standard for human antithrombin III concentrate. The equivalence in International Units of the International Standard is stated by the World Health Organization.
Method Prepare 2 independent series of 3 or 4 dilutions in the range 1/75 to 1/200 from 1 IU/mL, for both the preparation to be examined and the reference preparation, using tris-EDTA BSA buffer solution pH 8.4 R containing 15 IU of heparin per millilitre.
Warm 200 µL of each dilution at 37 °C for 1-2 min. Add to each dilution 200 µL of a solution of bovine thrombin R containing 2 IU/mL in tris-EDTA BSA buffer solution pH 8.4 R. Mix and maintain at 37 °C for exactly 1 min. Add 500 µL of a suitable chromogenic substrate (for example, d-phenylalanyl-l-pipecolyl-l-arginine-4-nitroanilide, reconstituted in water R to give a solution containing 4 mmol/L and further diluted to a concentration suitable for the assay using tris-EDTA BSA buffer solution pH 8.4 R without albumin). Immediately start measurement of the change in absorbance at 405 nm (2.2.25), continuing the measurement for at least 30 s. Calculate the rate of change of absorbance (ΔA/min). (Alternatively, an end-point assay may be used by stopping the reaction with acetic acid and measuring the absorbance at 405 nm.)
The rate of change of absorbance (ΔA/min) is inversely proportional to antithrombin III activity.
Check the validity of the assay and calculate the potency of the test preparation by the usual statistical methods (5.3).
B4. Assay of human protein C
1. chromogenic assay
Human protein C is a vitamin K-dependent plasma protein that, upon activation to activated protein C (APC), can inhibit blood coagulation through the proteolytic cleavage of factors Va and VIIIa. Human protein C activity is estimated using a two-step method: in the 1st step, human protein C in the preparation is activated by a specific activator from snake venom; in the 2nd step, APC cleaves a specific chromogenic substrate to form a chromophore that can be quantified spectrophotometrically.
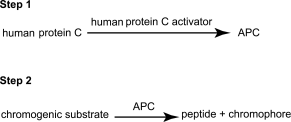
The potency of human protein C is estimated by comparing the ability of the preparation to be examined to cleave a chromogenic substrate with the same ability of a reference standard of human protein C calibrated in International Units. The International Unit is the activity of a stated amount of the International Standard for human protein C. The equivalence in International Units of the International Standard is stated by the World Health Organization.
Individual reagents may be obtained separately or in commercial kits. Both end-point and kinetic methods are available. Procedures and reagents may vary between different kits and the manufacturer’s instructions are followed. The essential features of the procedure are described in the following example of a microtitre plate end-point method.
REAGENTS
Dilution buffer pH 8.4 Dissolve 6.055 g of tris(hydroxymethyl)aminomethane R and 16.84 g of caesium chloride R in water R and adjust the pH if necessary. Dilute to 1000.0 mL with water R.
Human protein C activator Protein isolated from the venom of the viper Agkistrodon contortrix contortrix that specifically activates human protein C. Reconstitute and store according to the manufacturer’s instructions. Dilute to 0.25 U/mL with water R before use in the assay.
Activated protein C chromogenic substrate Specific chromogenic substrate for APC, for example l-pyroglutamyl-l-prolyl-l-arginyl-p-nitroaniline hydrochloride (pyroGlu-Pro-Arg-pNA.HCl). Reconstitute with water R to give a concentration of 4.5 mmol/L. Further dilute to 1.1 mmol/L with dilution buffer pH 8.4 before use in the assay.
METHOD
Reconstitute or thaw the preparation to be examined according to the manufacturer’s instructions. Dilute with water R to produce at least 3 separate dilutions for each preparation in the range 0.050-0.200 IU/mL, preferably in duplicate.
Step 1
Mix 0.025 mL of each dilution with 0.050 mL of the human protein C activator, both previously heated to 37 °C, and leave at 37 °C for exactly 10 min. For each dilution, prepare a blank in the same manner, using water R instead of the human protein C activator.
Step 2
Add 0.150 mL of diluted chromogenic substrate, previously heated to 37 °C, to each mixture and leave at 37 °C for exactly 10 min. The incubation time must be adjusted, if necessary, to ensure a linear development of chromophore with time. Terminate the reaction by adding 0.050 mL of a 50 per cent V/V solution of glacial acetic acid R.
Cleavage of the chromogenic substrate by APC causes release of the chromophore pNA, in proportion to the concentration of human protein C in the preparation. Measure the optical density at a wavelength of 405 nm. Subtract the optical density of the blank from the optical density of the test sample. Check the validity of the assay and calculate the potency of the preparation to be examined using the usual statistical methods (5.3).
2. clotting assay
Human protein C activity is estimated following cleavage to APC by a specific activator extracted from the venom of the viper Agkistrodon contortrix contortrix. The resulting APC inactivates factors Va and VIIIa, and thus prolongs the APTT (Activated Partial Thromboplastin Time) of a system in which all the coagulation factors are present, constant and in excess, except for human protein C, which is derived from the preparation being tested. Prolongation of the clotting time is proportional to the concentration of human protein C in the preparation.
The potency of human protein C is estimated by comparing the ability of the preparation to be examined to prolong the clotting time with the same ability of a reference standard of human protein C calibrated in International Units. The International Unit is the activity of a stated amount of the International Standard for human protein C. The equivalence in International Units of the International Standard is stated by the World Health Organization.
Individual reagents may be obtained separately or in commercial kits. Procedures and reagents may vary between different kits and the manufacturer’s instructions are followed. The essential features of the procedure are described in the following example.
REAGENTS
Dilution buffer pH 7.4 Isotonic non-chelating buffer.
Human protein C-deficient plasma Citrated human plasma with no measurable human protein C content. Reconstitute and store according to the manufacturer’s instructions.
Human protein C activator Protein isolated from the venom of the viper Agkistrodon contortrix contortrix that specifically activates human protein C. Reconstitute and store according to the manufacturer’s instructions.
Coagulation activator A suitable APTT reagent containing phospholipids and a contact activator may be used. It may be combined with the human protein C activator.
METHOD
Reconstitute or thaw the preparation to be examined according to the manufacturer’s instructions. Dilute with dilution buffer pH 7.4 to produce at least 3 separate dilutions for each preparation in the range 0.010-0.150 IU/mL, preferably in duplicate.
Mix 1 volume of each dilution with 1 volume of human protein C-deficient plasma and 1 volume of the human protein C activator (combined with the APTT reagent where appropriate), all previously heated to 37 °C. Add 1 volume of 0.025 M calcium chloride solution R previously heated to 37 °C, and record the clotting time.
The clotting time is proportional to the concentration of human protein C in each dilution. Check the validity of the assay and calculate the potency of the preparation to be examined using the usual statistical methods (5.3).
B5. Assay of human protein S
Human protein S is a vitamin K-dependent plasma protein that acts as a cofactor for the anticoagulant functions of activated protein C (APC). Human protein S activity may be determined by the clotting assay described below, which is sensitive to the ability of human protein S to accelerate the inactivation of factor Va by APC. In practice, the assay involves the addition of human protein S to a reagent mixture containing APC, factor Va and human protein S-deficient plasma. Prolongation of the clotting time is proportional to the concentration of human protein S in the preparation. Methods in which APC is added directly as a reagent are preferred to those in which APC is generated during the assay by the addition of a specific human protein C activator purified from snake venom. Activation of coagulation is initiated by the addition of an activating reagent such as thromboplastin or activated factor X, together with phospholipids and calcium chloride. During the assay, factor Va is generated from factor V in the human protein S-deficient plasma following the activation of coagulation. The assay procedure must ensure that human protein S is the only limiting factor.
The potency of human protein S is estimated by comparing the ability of the preparation to be examined to prolong the clotting time with the same ability of a reference standard of human protein S calibrated in International Units. The International Unit is the activity of a stated amount of the International Standard for human protein S. The equivalence in International Units of the International Standard is stated by the World Health Organization.
Individual reagents may be obtained separately or in commercial kits. Procedures and reagents may vary between different kits and the manufacturer′s instructions are followed. The essential features of the procedure are described in the following example.
REAGENTS
Dilution buffer pH 7.4 Isotonic non-chelating buffer prepared as follows: dissolve 6.08 g of tris(hydroxymethyl)aminomethane R and 8.77 g of sodium chloride R in water R and adjust the pH if necessary; add 10 g of bovine albumin R or human albumin R and dilute to 1000.0 mL with water R.
Human protein S-deficient plasma Citrated human plasma with no measurable human protein S content and, preferably, also free of C4b-binding protein.
Coagulation activator This reagent is used to initiate coagulation in the human protein S-deficient plasma, and thereby also provides a source of activated factor V. The activator may consist of tissue factor, activated factor X, or an agent capable of directly activating factor X that may be purified from the venom of Russell’s viper (Vipera russelli). The reagent also contains APC, phospholipids and calcium chloride R, or, alternatively, calcium chloride may be added separately after a timed activation period.
METHOD
Reconstitute or thaw the preparation to be examined according to the manufacturer’s instructions. Dilute with dilution buffer pH 7.4 to produce at least 3 separate dilutions for each preparation in the range 0.020-0.100 IU/mL, preferably in duplicate.
Mix 1 volume of each dilution with 1 volume of human protein S-deficient plasma, both previously heated to 37 °C. Add 2 volumes of the coagulation activator, previously heated to 37 °C, and record the clotting time.
Alternative procedures may use a coagulation activator without calcium chloride, and require a precisely timed activation period before the addition of calcium chloride and the measurement of clotting time.
The clotting time is proportional to the concentration of human protein S in each dilution. Check the validity of the assay and calculate the potency of the preparation to be examined using the usual statistical methods (5.3).
C1. Test for Fc function of immunoglobulin
REAGENTS
Stabilised human blood Collect group O human blood into ACD anticoagulant solution. Store the stabilised blood at 4 °C for not more than 3 weeks.
Phosphate-buffered saline pH 7.2 Dissolve 1.022 g of anhydrous disodium hydrogen phosphate R, 0.336 g of anhydrous sodium dihydrogen phosphate R and 8.766 g of sodium chloride R in 800 mL of water R and dilute to 1000 mL with the same solvent.
Magnesium and calcium stock solution Dissolve 1.103 g of calcium chloride R and 5.083 g of magnesium chloride R in water R and dilute to 25 mL with the same solvent.
Barbital buffer stock solution Dissolve 207.5 g of sodium chloride R and 25.48 g of barbital sodium R in 4000 mL of water R and adjust to pH 7.3 using 1 M hydrochloric acid. Add 12.5 mL of magnesium and calcium stock solution and dilute to 5000 mL with water R. Store at 4 °C in transparent containers.
Albumin barbital buffer solution Dissolve 0.150 g of bovine albumin R in 20 mL of barbital buffer stock solution and dilute to 100 mL with water R. Prepare immediately before use.
Tannic acid solution Dissolve 10 mg of tannic acid R in 100 mL of phosphate-buffered saline pH 7.2. Prepare immediately before use.
Guinea-pig complement Prepare a pool of serum from the blood of not fewer than 10 guinea-pigs. Separate the serum from the clotted blood by centrifugation at about 4 °C. Store the serum in small amounts below -70 °C. Immediately before starting complement-initiated haemolysis, dilute to 125-200 CH50 per millilitre with albumin barbital buffer solution and store in an ice-bath during the test.
Rubella antigen Suitable rubella antigen for haemagglutination-inhibition titre (HIT). Titre > 256 HA units.
Preparation of tanned human red blood cells
Separate human red blood cells by centrifuging an appropriate volume of stabilised human blood, wash the cells at least 3 times with phosphate-buffered saline pH 7.2 and suspend at 2 per cent V/V in phosphate-buffered saline pH 7.2. Add 0.2 mL of tannic acid solution to 14.8 mL of phosphate-buffered saline pH 7.2. Mix 1 volume of the freshly prepared dilution with 1 volume of the human red blood cell suspension and incubate at 37 °C for 10 min. Collect the cells by centrifugation (800 g for 10 min), discard the supernatant and wash the cells once with phosphate-buffered saline pH 7.2. Resuspend the tanned cells at 1 per cent V/V in phosphate-buffered saline pH 7.2.
Antigen coating of tanned human red blood cells
Take a suitable volume (Vs) of tanned cells, add 0.2 mL of rubella antigen per 1.0 mL of tanned cells and incubate at 37 °C for 30 min. Collect the cells by centrifugation (800 g for 10 min) and discard the supernatant. Add a volume of albumin barbital buffer solution equivalent to the discarded supernatant, resuspend and collect the cells as described and repeat the washing procedure. Resuspend with albumin barbital buffer solution using a volume equivalent to 3/4 of Vs, thereby obtaining the initial volume (Vi). Mix 900 µL of albumin barbital buffer solution with 100 µL of Vi, which is thereby reduced to the residual volume (Vr), and determine the initial absorbance at 541 nm (A). Dilute Vr by a factor equal to A using albumin barbital buffer solution, thereby obtaining the final adjusted volume Vf = Vr × A of sensitised human red blood cells and adjusting A to 1.0 ± 0.1 for a tenfold dilution.
Antibody binding of antigen-coated tanned human red blood cells
Prepare the following solutions in succession and in duplicate, using for each solution a separate half-micro cuvette (for example, disposable type) or test-tube.
(1) Test solutions. If necessary, adjust the immunoglobulin to be examined to pH 7.
Where method A is performed, dilute volumes of the preparation to be examined with albumin barbital buffer to obtain 30 mg and 40 mg of immunoglobulin and adjust the volume to 900 µL with albumin barbital buffer.
Where method B is performed, dilute volumes of the preparation to be examined with albumin barbital buffer to obtain 15 mg and 30 mg of immunoglobulin and adjust the volume to 1200 µL with albumin barbital buffer.
(2) Reference solutions. Prepare as for the test solutions using human immunoglobulin for Fc function BRP.
(3) Complement control. Albumin barbital buffer solution.
Where method A is performed, add to each cuvette/test-tube 100 µL of sensitised human red blood cells and mix well. Allow to stand for 15 min, add 1000 µL of albumin barbital buffer solution, collect the cells by centrifugation (1000 g for 10 min) of the cuvette/test-tube and remove 1900 µL of the supernatant. Replace the 1900 µL with albumin barbital buffer solution and repeat the whole of the washing procedure, finally leaving a volume of 200 µL. Test samples may be stored in sealed cuvettes/test-tubes at 4 °C for not longer than 24 h.
Where method B is performed, add to each test-tube 300 µL of sensitised human red blood cells and mix well (the final immunoglobulin concentration is in the range of 10-20 mg/mL). Allow to stand for 15 min, add 1500 µL of albumin barbital buffer solution and stir gently until homogeneous. Collect the cells by centrifugation (1000 g for 10 min) of the test-tube, remove the supernatant and add approximately 3 mL of albumin barbital buffer solution. Repeat this operation up to 4 times in total, leaving a final volume of 300 µL. Test samples may be stored in sealed test-tubes at 4 °C for not longer than 24 h.
Complement-initiated haemolysis
To measure haemolysis where method A is performed, add 600 µL of albumin barbital buffer solution warmed to 37 °C to the test sample, resuspend the cells carefully by repeated pipetting (not fewer than 5 times) and place the cuvette in the thermostatted cuvette holder of a spectrophotometer. After 2 min, add 200 µL of diluted guinea-pig complement (125-200 CH50/mL), mix thoroughly by pipetting twice and start immediately after the second pipetting the time-dependent recording of absorbance at 541 nm, using albumin barbital buffer solution as the compensation liquid. Stop the measurement if absorbance as a function of time has clearly passed the inflexion point.
To measure haemolysis where method B is performed, add 900 µL of albumin barbital buffer solution warmed to 37 °C to each test-tube and resuspend the cells carefully by repeated pipetting (not fewer than 5 times). The microtitre plate must be prewarmed to 37 °C before starting the test. Transfer 240 µL of each solution into 4 microtitre plate wells then incubate the microplate at 37 °C for 6 min, stirring gently every 10 s. To each microtitre plate well add 60 µL of diluted guinea-pig complement (150 CH50/mL). Mix for 10 s and immediately start recording the absorbance at 541 nm at 37 °C, measuring every 20 s. Stop the measurement if the absorbance as a function of time has clearly passed the inflexion point.
Evaluation
For each cuvette/test-tube/well, determine the slope (S) of the haemolysis curve at the approximate inflexion point by segmenting the steepest section in suitable time intervals (for example, Δt = 1 min), and calculate S between adjacent intersection points, expressed as ΔA per minute. The largest value for S serves as Sexp. In addition, determine the absorbance at the start of measurement (As) by extrapolating the curve, which is almost linear and parallel to the time axis within the first few minutes. Correct Sexp using the expression:
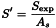
Calculate the arithmetic mean of the values of S′ for each preparation (test and reference solution). Calculate the index of Fc function (IFc) from the expression:
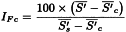
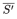 | = | arithmetic mean of the corrected slope for the preparation to be examined; |
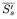 | = | arithmetic mean of the corrected slope for the reference preparation; |
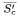 | = | arithmetic mean of the corrected slope for the complement control. |
Calculate the index of Fc function for the preparation to be examined: the value is not less than that stated in the leaflet accompanying the reference preparation.
C2. Test for anticomplementary activity of immunoglobulin
For the measurement of anticomplementary activity (ACA) of immunoglobulin, a defined amount of test material (10 mg of immunoglobulin) is incubated with a defined amount of guinea-pig complement (20 CH50) and the remaining complement is titrated; the anticomplementary activity is expressed as the percentage consumption of complement relative to the complement control considered as 100 per cent.
The haemolytic unit of complement activity (CH50) is the amount of complement that, in the given reaction conditions, will produce the lysis of 2.5 × 108 out of a total of 5 × 108 optimally sensitised red blood cells.
Magnesium and calcium stock solution Dissolve 1.103 g of calcium chloride R and 5.083 g of magnesium chloride R in water R and dilute to 25 mL with the same solvent.
Barbital buffer stock solution Dissolve 207.5 g of sodium chloride R and 25.48 g of barbital sodium R in 4000 mL of water R and adjust to pH 7.3 using 1 M hydrochloric acid. Add 12.5 mL of magnesium and calcium stock solution and dilute to 5000 mL with water R. Filter through a membrane filter (nominal pore size 0.22 µm). Store at 4 °C in glass containers.
Gelatin solution Dissolve 12.5 g of gelatin R in about 800 mL of water R and heat to boiling in a water-bath. Cool to 20 °C and dilute to 10 L with water R. Filter through a membrane filter (nominal pore size 0.22 µm). Store at 4 °C. Use clear solutions only.
Citrate solution Dissolve 8.0 g of sodium citrate R, 4.2 g of sodium chloride R and 20.5 g of glucose R in 750 mL of water R. Adjust to pH 6.1 using a 100 g/L solution of citric acid monohydrate R and dilute to 1000 mL with water R.
Gelatin barbital buffer solution Add 4 volumes of gelatin solution to 1 volume of barbital buffer stock solution and mix. Adjust to pH 7.3, if necessary, using 1 M sodium hydroxide or 1 M hydrochloric acid. Maintain at 4 °C. Prepare fresh solutions daily.
Stabilised sheep blood Collect 1 volume of sheep blood into 1 volume of citrate solution and mix. Store at 4 °C for not less than 7 days and not more than 28 days. (Stabilised sheep blood and sheep red blood cells are available from a number of commercial sources.)
Haemolysin Antiserum against sheep red blood cells prepared in rabbits. (Such antisera are available from a number of commercial sources.)
Guinea-pig complement Prepare a pool of serum from the blood of not fewer than 10 guinea-pigs. Separate the serum from the clotted blood by centrifugation at about 4 °C. Store the serum in small amounts below -70 °C.
METHOD
Preparation of standardised 5 per cent sheep red blood cell suspension
Separate sheep red blood cells by centrifuging an appropriate volume of stabilised sheep blood and wash the cells at least 3 times with gelatin barbital buffer solution and prepare a 5 per cent V/V suspension in the same solution. Measure the cell density of the suspension as follows: add 0.2 mL to 2.8 mL of water R and centrifuge the lysed solution for 5 min at 1000 g; the cell density is suitable if the absorbance (2.2.25) of the supernatant at 541 nm is 0.62 ± 0.01. Correct the cell density by adding gelatin barbital buffer solution according to the following equation:
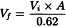
Vf | = | final adjusted volume; |
Vi | = | the initial volume; |
A | = | absorbance of the original suspension at 541 nm. |
The adjusted suspension contains about 1 × 109 cells/mL.
Haemolysin titration
Prepare haemolysin dilutions as shown in Table 2.6.17.-1.
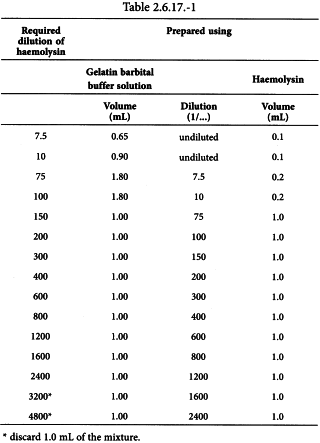
Add 1.0 mL of 5 per cent sheep red blood cell suspension to each tube of the haemolysin dilution series, starting at the 1/75 dilution, and mix. Incubate at 37 °C for 30 min.
Transfer 0.2 mL of each of these incubated mixtures to new tubes and add 1.10 mL of gelatin barbital buffer solution and 0.2 mL of diluted guinea-pig complement (for example, 1/150). Perform this in duplicate.
As the unhaemolysed cell control, prepare 3 tubes with 1.4 mL of gelatin barbital buffer solution and 0.1 mL of 5 per cent sheep red blood cell suspension.
As the fully haemolysed control, prepare 3 tubes with 1.4 mL of water R and 0.1 mL of 5 per cent sheep red cell suspension.
Incubate all tubes at 37 °C for 60 min and centrifuge at 1000 g for 5 min. Measure the absorbance (2.2.25) of the supernatants at 541 nm and calculate the percentage degree of haemolysis in each tube using the following expression:
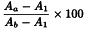
Aa | = | absorbance of tubes with haemolysin dilution; |
Ab | = | mean absorbance of the 3 tubes with full haemolysis; |
A1 | = | mean absorbance of the 3 tubes with no haemolysis. |
Plot the percentage degree of haemolysis as the ordinate against the corresponding reciprocal value of the haemolysin dilution as the abscissa on linear graph paper. Determine the optimal dilution of the haemolysin from the graph by inspection. Select a dilution such that further increase in the amount of haemolysin does not cause appreciable change in the degree of haemolysis. This dilution is defined as 1 minimal haemolytic unit (1 MHU) in 1.0 mL. The optimal haemolytic haemolysin dilution for preparation of sensitised sheep red blood cells contains 2 MHU/mL.
The haemolysin titration is not valid unless the maximum degree of haemolysis is 50 per cent to 70 per cent. If the maximum degree of haemolysis is not in this range, repeat the titration with more or less diluted complement solution.
Preparation of optimised sensitised sheep red blood cells (haemolytic system)
Prepare an appropriate volume of diluted haemolysin containing 2 MHU/mL and an equal volume of standardised 5 per cent sheep red blood cell suspension. Add the haemolysin dilution to the standardised cell suspension and mix. Incubate at 37 °C for 15 min, store at 2 °C to 8 °C and use within 6 h.
Titration of complement
Prepare an appropriate dilution of complement (for example 1/250) with gelatin barbital buffer solution and perform the titration in duplicate as shown in Table 2.6.17.-2.
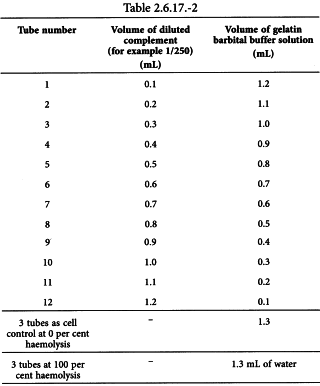
Add 0.2 mL of sensitised sheep red blood cells to each tube, mix well and incubate at 37 °C for 60 min. Cool the tubes in an ice-bath and centrifuge at 1000 g for 5 min. Measure the absorbance of the supernatant at 541 nm and calculate the degree of haemolysis (Y) using the following expression:
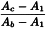
Ac | = | absorbance of tubes 1 to 12; |
Ab | = | mean absorbance of tubes with 100 per cent haemolysis; |
A1 | = | mean absorbance of cell controls with 0 per cent haemolysis. |
Plot Y/(1-Y) as the abscissa against the amount of diluted complement in millilitres as the ordinate on log–log graph paper. Fit the best line to the points and determine the ordinate for the 50 per cent haemolytic complement dose where Y/(1-Y) = 1.0. Calculate the activity in haemolytic units (CH50/mL) using the following expression:
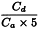
Cd | = | reciprocal value of the complement dilution; |
Ca | = | volume of diluted complement resulting in 50 per cent haemolysis, in millilitres; |
5 | = | scaling factor to take account of the number of red blood cells. |
The test is not valid unless the plot is a straight line between 15 per cent and 85 per cent haemolysis and the slope is 0.15 to 0.40, and preferably 0.18 to 0.30.
Test for anticomplementary activity
Prepare a complement dilution having 100 CH50/mL by diluting titrated guinea-pig complement with gelatin barbital buffer solution. Depending on the immunoglobulin to be examined and based on validation data, a pH adjustment to 7 may be necessary. Prepare incubation mixtures as follows for an immunoglobulin containing 50 mg/mL:

Carry out the test on the immunoglobulin to be examined and prepare ACA negative and positive controls using human immunoglobulin for anticomplementary activity BRP, as indicated in the leaflet accompanying the reference preparation. Higher or lower volumes of sample and of gelatin barbital buffer solution are added if the immunoglobulin concentration varies from 50 mg/mL; for example, 0.47 mL of gelatin barbital buffer solution is added to 0.33 mL of immunoglobulin containing 30 mg/mL to give 0.8 mL. Close the tubes and incubate at 37 °C for 60 min. Add 0.2 mL of each incubation mixture to 9.8 mL of gelatin barbital buffer solution to dilute the complement. Perform complement titrations on each tube as described above to determine the remaining complement activity (Table 2.6.17.-2). Calculate the anticomplementary activity of the preparation to be examined relative to the complement control considered as 100 per cent, using the following expression:
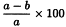
a | = | mean complement activity (CH50/mL) of complement control; |
b | = | complement activity (CH50/mL) of tested sample. |
The test is not valid unless:
C3. Assay of human anti-D immunoglobulin
METHOD A
The potency of human anti-D immunoglobulin is determined by comparing the quantity necessary to produce agglutination of D-positive red blood cells with the quantity of a reference preparation, calibrated in International Units, required to produce the same effect.
The International Unit is the activity contained in a stated amount of the International Reference Preparation. The equivalence in International Units of the International Reference Preparation is stated by the World Health Organization.
Human anti-D immunoglobulin BRP is calibrated in International Units by comparison with the International Standard and intended for use in the assay of human anti-D immunoglobulin.
Use pooled D-positive red blood cells, collected not more than 7 days earlier and suitably stored, obtained from not fewer than 4 group O R1R1 donors. To a suitable volume of the cells, previously washed 3 times with a 9 g/L solution of sodium chloride R, add an equal volume of bromelains solution R, allow to stand at 37 °C for 10 min, centrifuge, remove the supernatant and wash 3 times with a 9 g/L solution of sodium chloride R. Suspend 20 volumes of the red blood cells in a mixture of 15 volumes of inert serum, 20 volumes of a 300 g/L solution of bovine albumin R and 45 volumes of a 9 g/L solution of sodium chloride R. Stand the resulting suspension in iced water, stirring continuously.
Using a calibrated automated dilutor, prepare suitable dilutions of the preparation to be examined and of the reference preparation using as diluent a solution containing 5 g/L of bovine albumin R and 9 g/L of sodium chloride R.
Use a suitable apparatus for automatic continuous analysis. The following protocol is usually suitable: maintain the temperature in the manifold, except for the incubation coils, at 15.0 °C. Pump into the manifold of the apparatus the red blood cell suspension at a rate of 0.1 mL/min and a 3 g/L solution of methylcellulose 450 R at a rate of 0.05 mL/min. Introduce the dilutions of the preparation to be examined and the reference preparation at a rate of 0.1 mL/min for 2 min, followed by the diluent solution at a rate of 0.1 mL/min for 4 min before the next dilution is introduced.
Introduce air at a rate of 0.6 mL/min. Incubate at 37 °C for 18 min and then disperse the rouleaux by introducing at a rate of 1.6 mL/min a 9 g/L solution of sodium chloride R containing a suitable wetting agent (for example, polysorbate 20 R at a final concentration of 0.2 g/L) to prevent disruption of the bubble pattern. Allow the agglutinates to settle and decant twice, first at 0.4 mL/min and then at 0.6 mL/min. Lyse the unagglutinated red blood cells with a solution containing 5 g/L of octoxinol 10 R, 0.2 g/L of potassium ferricyanide R, 1 g/L of sodium hydrogen carbonate R and 0.05 g/L of potassium cyanide R at a rate of 2.5 mL/min. A 10-minute delay coil is introduced to allow for conversion of the haemoglobin. Continuously record the absorbance (2.2.25) of the haemolysate at a wavelength between 540 nm and 550 nm. Determine the range of antibody concentrations over which there is a linear relationship between the concentration and the resultant change in absorbance (ΔA). From the results, prepare a standard curve and use the linear portion of the curve to determine the activity of the preparation to be examined.
Calculate the potency of the preparation to be examined using the usual statistical methods (5.3).
METHOD B
The potency of human anti-D immunoglobulin is determined by competitive enzyme-linked immunoassay on erythrocyte-coated microtitre plates. The method is based on the competitive binding between a polyclonal anti-D immunoglobulin preparation and a biotinylated monoclonal anti-D antibody directed against a D-antigen-specific epitope. The activity of the preparation to be examined is compared with a reference preparation calibrated in International Units.
The International Unit is the activity of a stated amount of International Reference Preparation. The equivalence in International Units of the International Reference Preparation is stated by the World Health Organization.
Human anti-D immunoglobulin BRP is calibrated in International Units by comparison with the International Standard and intended for use in the assay of human anti-D immunoglobulin.
Materials
Reagents not specified are of analytical grade.
PBS (Phosphate-buffered saline) Dissolve 8.0 g of sodium chloride R, 0.76 g of anhydrous disodium hydrogen phosphate R, 0.2 g of potassium chloride R, 0.2 g of potassium dihydrogen phosphate R and 0.2 g of sodium azide R in water R and dilute to 1000 mL with the same solvent.
TBS (Tris-buffered saline) Dissolve 8.0 g of sodium chloride R and 0.6 g of tris(hydroxymethyl)aminomethane R in water R. Adjust to pH 7.2 with 1 M hydrochloric acid and dilute to 1000 mL with water R.
Papain solution Prepare a solution by stirring 1 g of papain R at 37 °C for 30 min in 10 mL of 0.067 M phosphate buffer solution pH 5.4 R, centrifuge at 10 000 g for 5 min and filter through a membrane filter (nominal pore size 0.22 µm). To activate, combine 1 mL of the filtrate with 1 mL of a 48.44 g/L solution of l-cysteine R and 1 mL of a 3.72 g/L solution of sodium edetate R and dilute to 10 mL with 0.067 M phosphate buffer solution pH 5.4 R. Freeze in aliquots at -20 °C or below.
Red blood cells Use pooled D-positive red blood cells obtained from not fewer than 3 group O R2R2 donors. Wash the cells 4 times with PBS. Centrifuge the cells at 1800 g for 5 min, mix a suitable volume of prewarmed packed cells with a suitable volume of prewarmed papain solution (2 volumes to 1 volume has been found suitable) and incubate at 37 °C for 10 min. Wash the cells 4 times with PBS. Store at 4 °C in an appropriate stabiliser for up to 1 week.
Biotinylated Brad-5 Use according to instructions.
Alkaline phosphatase-conjugated avidin/streptavidin reagent Preferably modified to combine high specific activity with low non-specific binding. Use according to instructions.
Substrate solution Use para-nitrophenyl phosphate according to instructions.
Cell fixation buffer Dissolve 18.02 g of glucose R, 4.09 g of sodium chloride R, 1.24 g of boric acid R, 10.29 g of sodium citrate R and 0.74 g of sodium edetate R in water R. Adjust to pH 7.2-7.3 using 1 M sodium hydroxide or 1 M hydrochloric acid, and dilute to 1000 mL with water R. Use directly from storage at 4 °C.
Glutaraldehyde solution Immediately before use, add 750 µL of a 250 g/L solution of glutaraldehyde R to 50 mL of cold PBS.
Microtitre plates Plates to be coated with red blood cells are flat-bottomed polystyrene plates with surface properties optimised for enzyme immunoassay and high protein-binding capacity. Plates used to prepare immunoglobulin dilutions are U- or V-bottomed polystyrene or poly(vinyl chloride) plates.
Method
Prepare a 0.1 per cent V/V suspension of papain-treated red blood cells in cold cell-fixation buffer. Pipette 50 µL into each well of the flat-bottomed microtitre plate.
Centrifuge the plate at 350 g for 3 min, preferably at 4 °C. Without removing the supernatant, gently add 100 µL of glutaraldehyde solution to each well and leave for 10 min.
Drain the wells by quickly inverting the plate and wash 3 times with 250-300 µL of PBS. This may be done manually or using a suitable automated plate washer. Either carry out the assay as described below, or store the plate at 4 °C after draining off the PBS and adding 100 µL of cell-fixation buffer per well and sealing with plastic film. Plates can be stored at 4 °C for up to 1 month.
Test solutions For freeze-dried preparations, reconstitute as stated on the label. Prepare 4 independent replicates of 5 serial 2-fold dilutions starting with 30 IU/mL in PBS containing 10 g/L of bovine albumin R. If necessary, adjust the starting dilution to obtain responses falling in the linear portion of the dose-response curve.
Reference solutions Reconstitute the reference preparation according to instructions. Prepare 4 independent replicates of 5 serial 2-fold dilutions starting with 30 IU/mL in PBS containing 10 g/L of bovine albumin R.
Using U- or V-bottomed microtitre plates, add 35 µL of each of the dilutions of the test solution or reference solution to each of a series of wells. To each well add 35 µL of biotinylated Brad-5 at 250 ng/mL.
Empty the wells of the red cell-coated plate by inverting and draining on a paper towel. Add 250 µL of PBS containing 20 g/L of bovine albumin R and leave at room temperature for 30 min.
Empty the wells of the red cell-coated plate by inverting and draining on a paper towel and transfer 50 µL from each of the dilutions of the test solution or reference solution containing biotinylated Brad-5 into the wells. Use 50 µL of PBS containing 10 g/L of bovine albumin R as negative control. Seal the plate with plastic film and incubate at room temperature for 1 h.
Remove the liquid from the wells of the red cell-coated plate and wash 3 times with 250-300 µL of TBS.
Dilute the alkaline phosphatase-conjugated avidin/streptavidin reagent in TBS containing 10 g/L of bovine albumin R and add 50 µL to each well. Incubate for 30 min at room temperature.
Remove the liquid from the wells of the red cell-coated plate and wash 3 times with 250-300 µL of TBS.
Add 100 µL of substrate solution to each of the wells and incubate at room temperature for 10 min in the dark. To stop the reaction, add 50 µL of 3 M sodium hydroxide to each of the wells.
Measure the absorbances at 405 nm and substract the negative control reading. Use the absorbance values in the linear range of the titration curve to estimate the potency of the preparation to be examined by the usual statistical methods (5.3).
METHOD C
The potency of human anti-D immunoglobulin is determined by flow cytometry in a microtitre plate format. The method is based on the specific binding between anti-D immunoglobulin and D-positive red blood cells. The activity of the preparation to be examined is compared with a reference preparation calibrated in International Units.
The International Unit is the activity of a stated amount of International Reference Preparation. The equivalence in International Units of the International Reference preparation is stated by the World Health Organization.
Human anti-D immunoglobulin BRP is calibrated in International Units by comparison with the International Standard and intended for use in the assay of human anti-D immunoglobulin.
Materials
Reagents not specified are of analytical grade.
PBS Dissolve 8.0 g of sodium chloride R, 0.76 g of disodium hydrogen phosphate dodecahydrate R, 0.2 g of potassium chloride R and 0.2 g of potassium dihydrogen phosphate R in water R and dilute to 1000 mL with the same solvent.
PBS-BSA solution PBS containing 10.0 g/L of bovine albumin R.
Red blood cells Use D-positive red blood cells obtained from a group O R1R1 donor within 2 weeks of collection. Store if necessary in an appropriate stabiliser at 4 °C. Wash the cells at least twice with PBS-BSA solution and prepare a suspension containing 1 × 104 cells per microlitre but not more than 5 × 104 cells per microlitre in PBS-BSA solution.
Use D-negative red blood cells obtained from a group O rr donor and prepared similarly.
Secondary antibody Use a suitable fluorescent dye-conjugated anti-IgG antibody fragment specific for human IgG or parts of it. Store and use according to the manufacturer’s instructions.
Microtitres plates Use flat-bottomed plates without surface treatment for enzyme immunoassays.
Method
Test solutions For freeze-dried preparations, reconstitute as stated on the label. Prepare at least 3 independent replicates of at least 3 serial 1.5- or 2-fold dilutions starting with a concentration in the range of 1.2-0.15 IU/mL using PBS/BSA solution as diluent. If necessary, adjust the starting dilution to obtain responses falling in the linear portion of the dose-response curve.
Reference solutions Reconstitute the reference preparation according to instructions. Prepare at least 3 independent replicates of at least 3 serial 1.5- or 2-fold dilutions starting with a concentration in the range of 1.2-0.15 IU/mL using PBS-BSA solution as diluent. If necessary, adjust the starting dilution to obtain responses falling in the linear portion of the dose-response curve.
Distribute 50 µL of the D-positive red blood cells into each well of a microtitre plate. Add 50 µL of each of the dilutions of the test solution or reference solution to each of a series of wells. Use 50 µL of PBS-BSA solution as negative control. Distribute 50 µL of the D-negative red blood cells into 4 wells of the same microtitre plate and add 50 µL of the lowest dilution of the test preparation. To monitor spurious reactions, distribute 50 µL of the D-positive red blood cells into 4 wells of the same microtitre plate and add 50 µL of PBS-BSA solution. Seal with plastic film and incubate at 37 °C for 40 min.
Centrifuge the plates at 50 g for 3 min, discard the supernatant and wash the cells with 200-250 µL of PBS-BSA solution. Repeat this at least once.
Centrifuge the plates at 50 g for 3 min, discard the supernatant and add 50 µL of the secondary antibody diluted with PBS-BSA solution to a suitable protein concentration. Seal with plastic film and incubate, protected from light, at room temperature for 20 min.
Centrifuge the plates at 50 g for 3 min, discard the supernatant and wash the cells with 200-250 µL of PBS-BSA solution. Repeat this at least once.
Centrifuge the plates at 50 g for 3 min, resuspend the cells into 200-250 µL of PBS. Transfer the cell suspension into a tube suitable for the flow-cytometry equipment available and further dilute by adding PBS to allow a suitable flow rate.
Proceed immediately with measurement of the median fluorescence intensity in a flow cytometer. Record at least 10 000 events without gating but excluding debris.
Use the median fluorescence intensity in the linear range of the dose-response curve to estimate the potency of the preparation to be examined by the usual statistical methods (5.3).
C4. Test for anti-D antibodies in human immunoglobulin
MATERIALS
Phosphate-buffered saline (PBS) Dissolve 8.0 g of sodium chloride R, 0.76 g of anhydrous disodium hydrogen phosphate R, 0.2 g of potassium chloride R and 0.2 g of potassium dihydrogen phosphate R in water R and dilute to 1000 mL with the same solvent. If the solution has to be kept for several days, 0.2 g of sodium azide R may be added in order to avoid microbial contamination.
PBS-BSA solution PBS containing 2 g/L of bovine albumin R (Cohn Fraction V, for ELISA). Store the solution at 2-8 °C but allow it to reach 19-25 °C before use.
Papain solution Use serological-grade papain from a commercial source, the activity of which has been validated.
Red blood cells Use pooled D-positive red blood cells from not fewer than 3 donors, preferably of group OR2R2. D-positive red blood cells may also be obtained from OR1R1 or OR1R2 donors. Mixing phenotypes has not been tested and is therefore not recommended.
Use pooled D-negative red blood cells, preferably from 3 donors of group Orr. When only 1 donor of group Orr is available, D-negative red blood cells from only 1 donor may be used.
Wash the cells 4 times with PBS or until the supernatant is clear. Each wash consists of suspending the cells in a minimum of 2 volumes of PBS, centrifuging the cells at 1800 g for 5 min to pack, and discarding the supernatant. Treat the packed cells with papain solution according to the manufacturer’s instructions and wash the cells 4 times with PBS.
Red blood cells may be stored for not more than 1 week in a preservative solution at 2-8 °C. A preparation of the following composition is appropriate:

If using stored cells, wash the cells at least twice in PBS or until the supernatant is clear before proceeding.
Microtitre plates Polystyrene plates from Greiner, catalogue No. 651101, are suitable. Use V-bottomed rigid microtitre plates.
Reference standards Immunoglobulin (anti-D antibodies test) BRP and Immunoglobulin (anti-D antibodies test negative control) BRP are suitable for use as the positive control and negative control, respectively.
METHOD
The test described in this chapter is performed at room temperature on the positive control solutions, the negative control solutions and the test solutions at the same time and under identical conditions.
Reference solutions Reconstitute the positive control and the negative control according to the instructions. The immunoglobulin G (IgG) concentration is 50 g/L in each of the reconstituted preparations. Make a 2-fold dilution of each reconstituted preparation with PBS-BSA solution to obtain solutions containing IgG at 25 g/L. Prepare 7 further serial 2-fold dilutions of each preparation using PBS-BSA solution to extend the dilution range to 1/256 (0.195 g/L IgG). Add 20 µL of each dilution of each preparation in duplicate to the microtitre plate.
Test solutions Dilute the preparation to be examined with PBS-BSA solution to obtain a starting IgG concentration of 25 g/L. For 50 g/L preparations, this is a 2-fold dilution; adjust the dilution factor accordingly for preparations with an IgG concentration other than 50 g/L to obtain a starting concentration of 25 g/L for testing. This 25 g/L solution is assigned a nominal 2-fold dilution factor for comparison with the reference solutions, even if this does not reflect the true dilution factor used to achieve 25 g/L. Prepare 7 further serial 2-fold dilutions of the preparation using PBS-BSA solution to extend the nominal dilution range to 1/256 (0.195 g/L IgG) for comparison with the reference preparations over the same IgG concentration range. Add 20 µL of each dilution in duplicate to the microtitre plate.
Prepare 3 per cent V/V suspensions of papain-treated D-positive (preferably OR2R2, but OR1R1 or OR1R2 may also be used) and D-negative (Orr) red blood cells in PBS-BSA solution. Add 20 µL of D-positive red blood cells to 1 dilution series of each of the preparation to be examined, the positive control and the negative control, and 20 µL of D-negative red blood cells to the other dilution series. Mix by shaking the plate on a shaker for 10 s (or until the cells are resuspended).
Centrifuge the plate at 80 g at room temperature for 1 min to pack the cells. Place the plate at an angle of approximately 70°. Read after 4-5 min or when the negative controls (D-negative red blood cells and negative control solution) have streamed. A cell button at the bottom of the well indicates a positive result. A stream of cells represents a negative result.
Record the endpoint titre as the reciprocal of the highest dilution that gives rise to a positive result.
The positive control has a nominal titre of 8 and the negative controls (D-negative red blood cells and negative control solution) must not show agglutination at the starting dilution of 1 in 2. Users must validate their own test conditions, and investigate their assay conditions and reagents in the event of results being significantly different from those expected. Failure to obtain negative reactions with the negative controls may indicate that, for example, insufficient time has elapsed for the cells to stream, or that reagents have been used directly from cold storage.
The titre of the preparation to be examined must not be greater than the titre of the positive control when both preparations are titrated from 25 g/L.
C5. Anti-A and anti-B haemagglutinins
METHOD a: INDIRECT METHOD
Prepare in duplicate serial dilutions of the preparation to be examined in a 9 g/L solution of sodium chloride R. To each dilution of 1 series add an equal volume of a 5 per cent V/V suspension of group A1 red blood cells previously washed 3 times with the sodium chloride solution. To each dilution of the other series add an equal volume of a 5 per cent V/V suspension of group B red blood cells previously washed 3 times with the sodium chloride solution. Incubate the suspensions at 37 °C for 30 min then wash the cells 3 times with the sodium chloride solution. Leave the cells in contact with a polyvalent anti-human globulin reagent for 30 min. Without centrifuging, examine each suspension for agglutination under a microscope.
METHOD B: DIRECT METHOD
MATERIALS
Phosphate-buffered saline (PBS) Dissolve 8.0 g of sodium chloride R, 0.76 g of anhydrous disodium hydrogen phosphate R, 0.2 g of potassium chloride R and 0.2 g of potassium dihydrogen phosphate R in water R and dilute to 1000 mL with the same solvent. If the solution has to be kept for several days, 0.2 g of sodium azide R may be added in order to avoid microbial contamination.
PBS-BSA solution PBS containing 2 g/L of bovine albumin R (Cohn Fraction V, for ELISA). Store the solution at 2-8 °C but allow it to reach 19-25°C before use.
Papain solution Use serological-grade papain from a commercial source, the activity of which has been validated.
Red blood cells Use pooled D-negative A1 (A1rr), D-negative B (Brr) and D-negative O (Orr) red blood cells from preferably 3 donors. When Immunoglobulin for anti-A and anti-B antibodies limit test BRP is used, 3 donors are to be used. A2 red blood cells are not recommended as they give weaker reactions.
Wash the cells 4 times with PBS or until the supernatant is clear. Each wash consists of suspending the cells in a minimum of 2 volumes of PBS, centrifuging the cells at 1800 g for 5 min to pack, and discarding the supernatant. Treat the packed cells with papain solution according to the manufacturer’s instructions and wash the cells 4 times with PBS.
Red blood cells may be stored for not more than 1 week in a preservative solution at 2-8 °C. A preparation of the following composition is appropriate:

If using stored cells, wash the cells at least twice in PBS or until the supernatant is clear before proceeding.
Microtitre plates Use V-bottomed rigid microtitre plates.
Reference standards Immunoglobulin (anti-A, anti-B antibodies test positive control) BRP and Immunoglobulin (anti-A, anti-B antibodies test negative control) BRP are suitable for use as the positive control and negative control, respectively, and should be used as guides for operators establishing and performing the direct method for anti-A and anti-B haemagglutinins.
Immunoglobulin for anti-A and anti-B antibodies limit test BRP defines the recommended maximum limits permissible for batches of human immunoglobulin and must be used only for comparison with batches of human immunoglobulin that have higher titres than the positive control.
METHOD
The test described in this chapter is performed at room temperature on the positive control solutions, the negative control solutions and the test solutions at the same time and under identical conditions. Whenever necessary, a further test is performed with Immunoglobulin for anti-A and anti-B antibodies limit test BRP.
Reference solutions Reconstitute the positive control and the negative control according to the instructions. The immunoglobulin G (IgG) concentration is 50 g/L in each of the reconstituted preparations. Make a 2-fold dilution of each reconstituted preparation with PBS-BSA solution to obtain solutions containing IgG at 25 g/L. Prepare 7 further serial 2-fold dilutions of each preparation using PBS-BSA solution to extend the dilution range to 1/256 (0.195 g/L IgG). Add 20 µL of each dilution of each preparation in triplicate to the microtitre plate.
Test solutions Dilute the preparation to be examined with PBS-BSA solution to obtain a starting IgG concentration of 25 g/L. For 50 g/L preparations, this is a 2-fold dilution; adjust the dilution factor accordingly for preparations with an IgG concentration other than 50 g/L to obtain a starting concentration of 25 g/L for testing. This 25 g/L solution is assigned a nominal 2-fold dilution factor for comparison with the reference solutions, even if this does not reflect the true dilution factor used to achieve 25 g/L. Prepare 7 further serial 2-fold dilutions of the preparation using PBS-BSA solution to extend the nominal dilution range to 1/256 (0.195 g/L IgG) for comparison with the reference preparations over the same IgG concentration range. Add 20 µL of each dilution in triplicate to the microtitre plate.
Prepare 3 per cent V/V suspensions of papain-treated D-negative A1, B and O red blood cells in PBS/BSA solution. Add 20 µL of D-negative A1, B and O red blood cells respectively to the 1st, the 2nd and the 3rd dilution series of each of the preparation to be examined, the positive control and the negative control. Mix by shaking the plate on a shaker for 10 s (or until the cells are resuspended).
Centrifuge the plate at 80 g at room temperature for 1 min to pack the cells. Place the plate at an angle of approximately 70°. Read after 4-5 min or when the negative controls (D-negative O red blood cells and negative control solution) have streamed. A cell button at the bottom of the well indicates a positive result. A stream of cells represents a negative result.
Record the endpoint titre as the reciprocal of the highest dilution that gives rise to a positive result.
The positive control has nominal anti-A and anti-B titres of 32 (range 32-64 for anti-A; range 16-32 for anti-B) and the negative controls (D-negative O red blood cells and negative control solution) must not show agglutination at the starting dilution of 1 in 2. Users must validate their own test conditions, and investigate their assay conditions and reagents in the event of results being significantly different from those expected. Failure to obtain negative reactions with the negative controls may indicate that, for example, insufficient time has elapsed for the cells to stream, or that reagents have been used directly from cold storage.
If the anti-A or anti-B titre of the preparation to be examined is greater than the titre of the positive control when both preparations are titrated from 25 g/L, the test preparation is to be compared with Immunoglobulin for anti-A and anti-B antibodies limit test BRP.
The maximum allowable titre is 64 when the preparations are titrated from 25 g/L.
Other blood-related components
D1. Assay of human α-1-proteinase inhibitor
Human α-1-proteinase inhibitor (also known as α-1-antitrypsin or α-1-antiproteinase) content is determined by comparing the ability of the preparation to be examined to inactivate the serine protease elastase (porcine pancreatic elastase or human neutrophil elastase) with the same ability of a reference standard of human α-1-proteinase inhibitor calibrated in milligrams of active (functional) α-1-proteinase inhibitor. Varying quantities of the preparation to be examined are mixed with a given quantity of elastase and the remaining elastase activity is determined using a suitable chromogenic substrate. The method described below is given as an example.
Reagents
Tris-albumin buffer solution Dissolve 24.23 g of trometamol R in water R, adjust to pH 8.0 ± 0.3 using hydrochloric acid R1 and dilute to 1000 mL with water R. To 100 mL of this solution add 0.5 mL of a 20 per cent solution of human albumin R or bovine albumin R.
Buffer solution containing human or bovine albumin must be prepared fresh on the day of its use; otherwise, it can be conserved by sterile filtration (0.2 µm) and stored at 2-8 °C for up to 2 weeks.
METHOD
Prepare 2 series of 4 or 5 dilutions in an appropriate human α-1-proteinase inhibitor concentration range, for both the preparation to be examined and the reference standard, using the tris-albumin buffer solution.
Transfer 50 µL of the reference solution dilutions into the wells of a microtitre plate and to each well, add 150 µL of a porcine pancreatic elastase solution diluted to an appropriate concentration with the tris-albumin buffer solution. Incubate for a defined period of time, 3-10 min, at room temperature. Since the activity of the solutions of the different porcine pancreatic elastases may vary, the concentration of elastase can be adjusted by evaluation of blank values containing elastase but no human α-1-proteinase inhibitor, to exhibit a suitable change of absorbance at 405 nm under the actual assay conditions.
Add to each well 100 µL of a solution of chromogenic substrate N-succinyl-tri-l-alanyl 4-p-nitroanilide (Suc-Ala-Ala-Ala-pNA), reconstituted in dimethyl sulfoxide R to give a solution containing 4.5 mg/mL, then further diluted with the tris-albumin buffer solution to a concentration of 0.45 mg/mL. Immediately start measurement of the change in absorbance (2.2.25) at 405 nm using a microtitre plate reader, continuing the measurement for at least 5 min. Calculate the rate of change of absorbance (ΔA/min). Alternatively, an end-point assay may be used by stopping the reaction with acetic acid and measuring the absorbance at 405 nm. If the assay is performed in test tubes using spectrophotometers for monitoring the change in absorbance at 405 nm, the volumes of reagent solutions are changed proportionally.
The rate of change of absorbance (ΔA/min) is inversely proportional to human α-1-proteinase inhibitor activity.
Check the validity of the assay and calculate the potency of the test preparation by the usual statistical methods (5.3).
D2. Assay of human C1-esterase inhibitor
The human C1-esterase inhibitor content of the preparation to be examined is determined by comparing its ability to inhibit C1-esterase with that of a reference preparation calibrated in International Units. The International Unit is the activity of a stated amount of the International Standard for human plasma-derived C1-esterase inhibitor concentrate. The equivalence in International Units of the International Standard is stated by the World Health Organization. Varying quantities of the preparation to be examined are mixed with an excess of C1-esterase and the remaining C1-esterase activity is determined using a suitable chromogenic substrate.
Individual reagents may be obtained separately or in commercial kits. Procedures and reagents may vary between different kits and the manufacturer’s instructions are followed. The essential features of the procedure are described in the following example of a microtitre-plate kinetic method.
Reconstitute the preparation as stated on the label. Prepare an appropriate series of at least 3 dilutions, from 1 IU/mL of C1-esterase inhibitor, for both the preparation to be examined and the reference preparation, using a suitable pH 7.4 buffer solution containing 9 g/L of sodium chloride R and either 10 g/L of human albumin R or 10 g/L of bovine albumin R. Warm all solutions to 37 °C. Place a suitable quantity of 1 of the dilutions of the reference preparation or of the preparation to be examined in microtitre plate wells and incubate at 37 °C. To each well add a suitable quantity of C1-esterase solution and incubate at 37 °C for 5 min. Add an appropriate quantity of a suitable specific chromogenic substrate such as methoxycarbonyl-L-lysyl(ε-benzyloxycarbonyl)-glycyl-L-arginine-4-nitroanilide. Read the rate of increase of absorbance (ΔA/min) at 405 nm. A positive control, using the pH 7.4 buffer solution instead of the C1-esterase inhibitor, is included. For preparations that may exhibit proteolytic activity, a test is carried out on a suitable blank composed of the preparation to be examined, the pH 7.4 buffer solution and the chromogenic substrate.
Calculate the C1-esterase inhibitor content using the usual statistical methods, for example slope ratio (5.3).