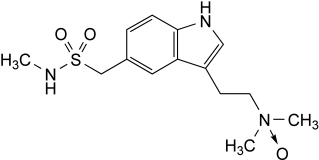
Sumatriptan Succinate
Action and use
Serotonin 5HT1 receptor agonist; treatment of migraine.
Preparations
Ph Eur
DEFINITION
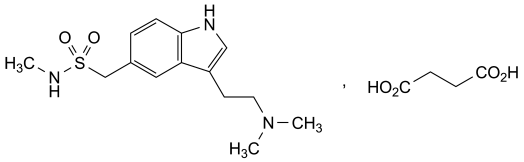
[3-[2-(Dimethylamino)ethyl]-1H-indol-5-yl]-N-methylmethanesulfonamide hydrogen butanedioate.
Content
97.5 per cent to 102.0 per cent (anhydrous substance).
CHARACTERS
Appearance
White or almost white powder.
Solubility
Freely soluble in water, sparingly soluble in methanol, practically insoluble in methylene chloride.
IDENTIFICATION
Infrared absorption spectrophotometry (2.2.24).
Comparisonsumatriptan succinate CRS.
TESTS
Solution S
Dissolve 1.0 g in carbon dioxide-free water R and dilute to 25.0 mL with the same solvent.
pH (2.2.3)
4.5 to 5.3.
Dilute 2.5 mL of solution S to 10 mL with carbon dioxide-free water R.
Absorbance (2.2.25)
Maximum 0.10, determined at 440 nm on solution S.
Impurities A and H
Liquid chromatography (2.2.29).
Test solution Dissolve 30.0 mg of the substance to be examined in the mobile phase and dilute to 10.0 mL with the mobile phase.
Reference solution (a) Dilute 1.0 mL of the test solution to 100.0 mL with the mobile phase. Dilute 1.0 mL of this solution to 10.0 mL with the mobile phase.
Reference solution (b) Dissolve 3 mg of sumatriptan for system suitability CRS (containing impurities A and H) in the mobile phase and dilute to 1.0 mL with the mobile phase.
Mobile phase Mix 10 volumes of a 771 g/L solution of ammonium acetate R and 90 volumes of methanol R.
Flow rate 2.0 mL/min.
Detection Spectrophotometer at 282 nm.
Injection 20 µL.
Run time 5 times the retention time of sumatriptan.
Identification of impurities Use the chromatogram supplied with sumatriptan for system suitability CRS and the chromatogram obtained with reference solution (b) to identify the peaks due to impurities A and H.
Relative retention With reference to sumatriptan (retention time = about 2 min): impurity A = about 1.9; impurity H = about 2.6.
System suitability: reference solution (b):
Related substances
Liquid chromatography (2.2.29).
Solution A Dissolve 2.925 g of sodium dihydrogen phosphate R in 600 mL of water R, adjust to pH 6.5 with strong sodium hydroxide solution R, dilute to 750 mL with water R, add 250 mL of acetonitrile R and mix.
Test solution (a) Dissolve 30.0 mg of the substance to be examined in the mobile phase and dilute to 10.0 mL with the mobile phase.
Test solution (b) Dissolve 15.0 mg of the substance to be examined in solution A and dilute to 100.0 mL with solution A.
Reference solution (a) Dilute 1.0 mL of test solution (a) to 100.0 mL with the mobile phase. Dilute 1.0 mL of this solution to 10.0 mL with the mobile phase.
Reference solution (b) Dissolve the contents of a vial of sumatriptan impurity mixture CRS (containing impurities B, C, D and E) in the mobile phase and dilute to 1 mL with the mobile phase.
Reference solution (c) Dissolve 15.0 mg of sumatriptan succinate CRS in solution A and dilute to 100.0 mL with solution A.
Mobile phase Mix 25 volumes of acetonitrile R with 75 volumes of a solution prepared as follows: dissolve 0.970 g of dibutylamine R, 0.735 g of phosphoric acid R and 2.93 g of sodium dihydrogen phosphate R in 750 mL of water R, adjust to pH 6.5 with strong sodium hydroxide solution R and dilute to 1000 mL with water R.
Flow rate 1.5 mL/min.
Detection Spectrophotometer at 282 nm.
Injection 10 µL of test solution (a) and reference solutions (a) and (b).
Run time 4 times the retention time of sumatriptan.
Identification of impurities Use the chromatogram supplied with sumatriptan impurity mixture CRS and the chromatogram obtained with reference solution (b) to identify the peaks due to impurities B, C, D and E.
Relative retention With reference to sumatriptan (retention time = about 7 min): impurity E = about 0.5; impurity B = about 0.6; impurity D = about 0.7; impurity C = about 0.8.
Water (2.5.12)
Maximum 1.0 per cent, determined on 0.500 g.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
Liquid chromatography (2.2.29) as described in the test for related substances with the following modification.
Injection Test solution (b) and reference solution (c).
Calculate the percentage content of C18H27N3O6S taking into account the assigned content of sumatriptan succinate CRS.
STORAGE
Protected from light.
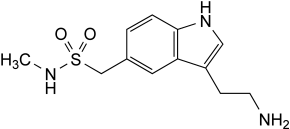
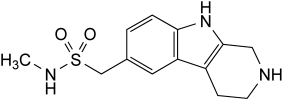
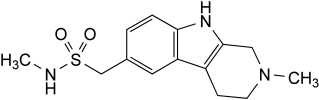
IMPURITIES
Specified impurities A, B, C, D, E, H
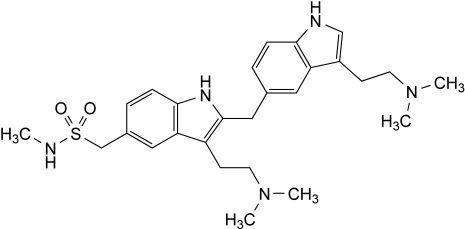
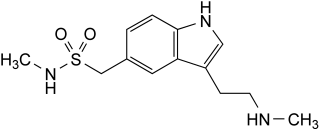
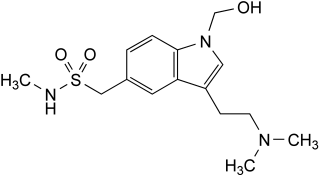
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use): F, G.
Ph Eur