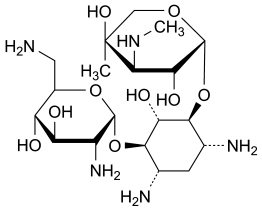
Gentamicin Sulfate
Action and use
Aminoglycoside antibacterial.
Preparations
Gentamicin and Hydrocortisone Acetate Ear Drops
Ph Eur
DEFINITION
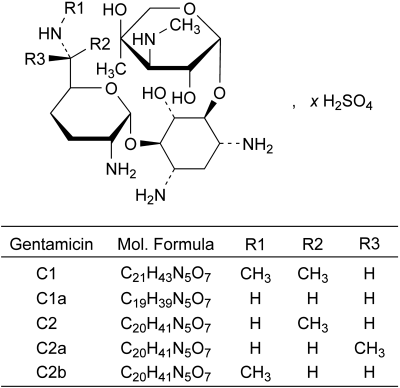
Mixture of the sulfates of antimicrobial substances produced by Micromonospora purpurea, the main components being gentamicins C1, C1a, C2, C2a and C2b.
Content
Minimum 590 IU/mg (anhydrous substance).
CHARACTERS
Appearance
White or almost white, hygroscopic powder.
Solubility
Freely soluble in water, practically insoluble in ethanol (96 per cent).
IDENTIFICATION
First identification: B, C
Second identification: A, C
Test solution Dissolve 25 mg of the substance to be examined in water R and dilute to 5 mL with the same solvent.
Reference solution Dissolve the contents of a vial of gentamicin sulfate CRS in water R and dilute to 5 mL with the same solvent.
PlateTLC silica gel plate R.
Mobile phase The lower layer of a mixture of equal volumes of concentrated ammonia R, methanol R and methylene chloride R.
Application 10 µL.
Development Over 2/3 of the plate.
Drying In air.
Detection Spray with ninhydrin solution R1 and heat at 110 °C for 5 min.
Results The 3 principal spots in the chromatogram obtained with the test solution are similar in position, colour and size to the 3 principal spots in the chromatogram obtained with the reference solution.
Results The chromatogram obtained with test solution (b) shows 5 principal peaks having the same retention times as the 5 principal peaks in the chromatogram obtained with reference solution (a).
TESTS
Solution S
Dissolve 0.8 g in carbon dioxide-free water R and dilute to 20 mL with the same solvent.
Appearance of solution
Solution S is clear (2.2.1) and not more intensely coloured than intensity 6 of the range of reference solutions of the most appropriate colour (2.2.2, Method II).
pH (2.2.3)
3.5 to 5.5 for solution S.
Specific optical rotation (2.2.7)
+ 107 to + 121 (anhydrous substance).
Dissolve 2.5 g in water R and dilute to 25.0 mL with the same solvent.
Composition
Liquid chromatography (2.2.29): use the normalisation procedure taking into account only the peaks due to gentamicins C1, C1a, C2, C2a and C2b.
Test solution (a) Dissolve 25.0 mg of the substance to be examined in the mobile phase and dilute to 25.0 mL with the mobile phase.
Test solution (b) Dilute 5.0 mL of test solution (a) to 25.0 mL with the mobile phase.
Reference solution (a) Dissolve 5 mg of gentamicin for peak identification CRS (containing impurity B) in the mobile phase and dilute to 25 mL with the mobile phase.
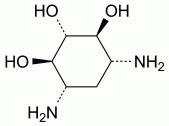
Reference solution (b) Dissolve 20.0 mg of sisomicin sulfate CRS (impurity A) in the mobile phase and dilute to 20.0 mL with the mobile phase.
Reference solution (c) Dilute 1.0 mL of reference solution (b) to 100.0 mL with the mobile phase.
Reference solution (d) To 1 mL of reference solution (b), add 5 mL of test solution (a) and dilute to 50 mL with the mobile phase.
Mobile phase To 900 mL of carbon dioxide-free water R, add 7.0 mL of trifluoroacetic acid R, 250.0 µL of pentafluoropropanoic acid R and 4.0 mL of carbonate-free sodium hydroxide solution R, allow to equilibrate and adjust to pH 2.6 using carbonate-free sodium hydroxide solution R diluted 1 to 25. Add 15 mL of acetonitrile R and dilute to 1000.0 mL with carbon dioxide-free water R.
Flow rate 1.0 mL/min.
Post-column solutioncarbonate-free sodium hydroxide solution R diluted 1 to 25, previously degassed, which is added pulse-less to the column effluent using a 375 µL polymeric mixing coil.
Flow rate of post-column solution 0.3 mL/min.
Detection Pulsed amperometric detector or equivalent with a gold indicator electrode, a silver-silver chloride reference electrode, and a stainless steel auxiliary electrode which is the cell body, held at respectively + 0.05 V detection, + 0.75 V oxidation and -0.15 V reduction potentials, with pulse durations according to the instrument used.
Injection 20 µL of test solution (b) and reference solutions (a), (c) and (d).
Run time 1.2 times the retention time of gentamicin C1.
Identification of peaks Use the chromatogram supplied with gentamicin for peak identification CRS to identify the peaks due to gentamicins C1, C1a, C2, C2a and C2b.
Relative retention With reference to impurity A (retention time = about 23 min): gentamicin C1a = about 1.1; gentamicin C2 = about 1.8; gentamicin C2b = about 2.0; gentamicin C2a = about 2.3; gentamicin C1 = about 3.0.
Related substances
Liquid chromatography (2.2.29) as described in the test for composition with the following modifications; use reference solution (c) to calculate the percentage content of each impurity.
Injection 20 µL of test solution (a) and reference solutions (a) and (c).
Identification of impurities Use the chromatogram obtained with reference solution (c) to identify the peak due to impurity A; use the chromatogram supplied with gentamicin for peak identification CRS and the chromatogram obtained with reference solution (a) to identify the peak due to impurity B.
Methanol (2.4.24, System B)
Maximum 1.0 per cent.
Sulfate
32.0 per cent to 35.0 per cent (anhydrous substance).
Dissolve 0.250 g in 100 mL of distilled water R and adjust the solution to pH 11 using concentrated ammonia R. Add 10.0 mL of 0.1 M barium chloride and about 0.5 mg of phthalein purple R. Titrate with 0.1 M sodium edetate, adding 50 mL of ethanol (96 per cent) R when the colour of the solution begins to change and continue the titration until the violet-blue colour disappears.
1 mL of 0.1 M barium chloride is equivalent to 9.606 mg of SO4.
Water (2.5.12)
Maximum 15.0 per cent, determined on 0.300 g.
Sulfated ash (2.4.14)
Maximum 1.0 per cent, determined on 0.50 g.
Bacterial endotoxins (2.6.14)
Less than 0.71 IU/mg, if intended for use in the manufacture of parenteral preparations without a further appropriate procedure for the removal of bacterial endotoxins.
ASSAY
Carry out the microbiological assay of antibiotics (2.7.2).
STORAGE
In an airtight container. If the substance is sterile, store in a sterile, airtight, tamper-proof container.
IMPURITIES
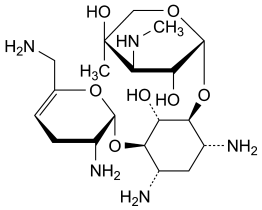
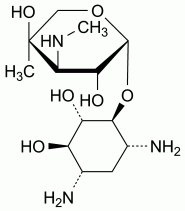
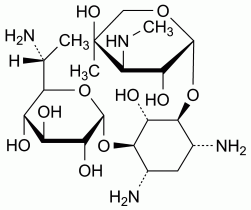
Specified impurities A, B
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use): C, D, E.
Ph Eur