SC I E. Dissolution Testing of Solid Oral Dosage Forms
This section provides information on the pharmacopoeial dissolution test and guidance on its function and application in individual monographs of the British Pharmacopoeia for tablets and capsules.
1. Harmonisation
1.1 The dissolution test for solid oral dosage forms in Appendix XII B1 of the British Pharmacopoeia is that of the European Pharmacopoeia (Ph. Eur. method 2.9.3). It is a harmonised text prepared by the Pharmacopoeial Discussion Group (PDG). The PDG is comprised of representatives of the European Pharmacopoeia, Japanese Pharmacopoeia and the United States Pharmacopeia. Although the test is largely harmonised, some regional differences remain.
1.2 It is not the intention of the British Pharmacopoeia to apply retrospectively the harmonised test conditions and acceptance criteria to BP monographs or to change unilaterally specifications for existing products. Appendix XII B1 contains a section entitled Monographs of the British Pharmacopoeia which provides requirements for monographs for tablets and capsules of the BP published prior to 2008. Taking account of permissible assay ranges and content uniformity, this pharmacopoeial (that is, shelf-life) dissolution requirement is considered to offer an acceptable degree of assurance of ‘complete dissolution’. The choice of a time is, of necessity, somewhat arbitrary but 45 minutes is considered satisfactory for the majority of conventional-release (non-modified-release) products.
2. Apparatus
2.1 Four types of apparatuses are now described in the British and European Pharmacopoeias; the basket, the paddle, the reciprocating cylinder and the flow-through cell. The descriptions are concordant with those published in the United States Pharmacopeia (USP).
2.2 Of the two established apparatuses (basket and paddle) the paddle is now the apparatus of choice for many preparations. However, where a published test uses the basket, work to validate a change to the paddle method is not contemplated. The reciprocating cylinder is useful for pH profiling studies while the flow-through cell may be appropriate for preparations of poorly soluble active ingredients (see Appendix XII B Annex).
3. Test conditions and acceptance criteria
3.1 Test conditions The harmonised test conditions included in Appendix XII B1 will be applied to all new monographs of the British Pharmacopoeia. It is not the intention of the British Pharmacopoeia Commission to apply these criteria retrospectively to existing monographs. Where an individual monograph prescribes the use of the requirements stated under Monographs of the British Pharmacopoeia in Appendix XII B1, the following conditions using the basket or paddle apparatus are preferred.
— rotation speed:100 rpm (basket), 50 rpm (paddle)
— dissolution medium volume: 900 mL
— dissolution medium composition: aqueous, commonly 0.1m hydrochloric acid or phosphate buffers of pH 6.8 to 7.6
— number of units tested: 6 (plus 6, if a retest is required).
— The number of units tested is specified in Appendix XII B1; other conditions are specified in the relevant individual monographs.
In situations where it has been demonstrated that the harmonised criteria are not applicable (e.g. low solubility preparations, ‘coning’ of material in the vessel, low concentration of analyte), modifications may be made to the test conditions, such as, adding a surfactant, increasing the paddle rotation speed or using a modified vessel and reducing the volume of dissolution medium used.
3.2 Acceptance criteria For new monographs for conventional-release preparations, unless otherwise stated, the harmonised acceptance criteria (“Q” values) in Appendix XII B1 should be applied. For monographs published prior to the BP 2008, the established BP criteria using either the basket or the paddle apparatus are specified under ‘Monographs of the British Pharmacopoeia’ in Appendix XII B1.
3.3 The schematic diagram outlines which limits to apply in each case (Fig. SCIE-1).
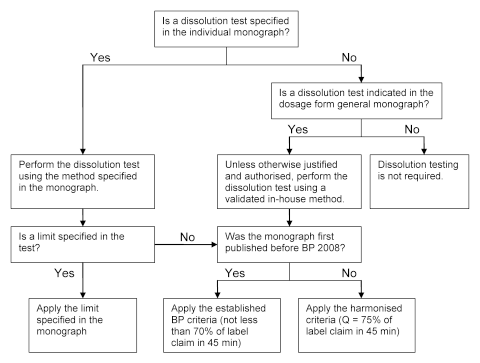
Figure SCIE-1
3.4 A list of those British Pharmacopoeia monographs that were published up to and including the BP 2007 is provided below.
Clofibrate Capsules | |
Desipramine Tablets | |
Iopanoic Acid Tablets | |
Tiabendazole Chewable Tablets | |
BP (Vet): | |
Acepromazine Tablets | Lincomycin Tablets |
Amoxicillin Tablets | Phenylbutazone Tablets |
Chlortetracycline Tablets | Piperazine Citrate Tablets |
Co-trimazine Tablets | |
Etamiphylline Tablets | |
Piperazine Adipate Tablets |
3.5 Standardised conditions and limits are considered appropriate for a pharmacopoeial test that is intended to apply to monographs covering products from different manufacturers. It might be argued that non-standardised conditions and limits would be more discriminatory but ‘tailor-made’ test conditions and limits may introduce product bias and may discriminate unnecessarily between products that are equally acceptable from a clinical view-point. Similarly with sufficient manipulation of the test conditions, dissolution of almost any product can be achieved. Ideally the test should reflect clinically significant differences in bioavailability arising from differences in dissolution in such a way that clinically acceptable formulations will pass whereas clinically unacceptable formulations will fail.
3.6 Another issue that has been considered in relation to test conditions and criteria is that of multiple-point dissolution profiles as opposed to single-point dissolution tests. It has been concluded that for conventional-release preparations such an extension of testing is not generally necessary or appropriate for pharmacopoeial purposes.
4. Function
4.1 The ultimate objective of dissolution testing may be described as ensuring adequate and reproducible bioavailability without recourse to routine in-vivo testing. On some occasions, this may be achieved by dissolution testing of a particular product for which in vitro/in vivo correlation has been demonstrated.
4.2 A more common objective of dissolution testing is to obtain information about the drug release characteristics of a particular formulation or batch of product under standardised in vitro testing conditions.
4.3 Dissolution testing may also be carried out during product development studies and is a useful tool in optimising formulation and manufacturing parameters. It is usually required by the Competent Authority as part of a marketing authorisation application.
4.4 Once a product is licensed, dissolution testing may be required routinely as part of quality control to demonstrate consistency of manufacture before the release of each batch of the finished product or, when necessary, to provide evidence to support changes in manufacture such as minor changes in formulation or process, changes in site or changes in immediate packaging materials.
5. Application
5.1 The British Pharmacopoeia Commission has not adopted a policy of universal application of the dissolution test and, therefore, a dissolution requirement will not be included automatically in every capsule and tablet monograph. The International Conference on Harmonisation (ICH) guideline on Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products (ICH Q6A) contains guidance on the application of dissolution testing to drug products and this will be used as a basis for deciding whether to include a dissolution test in an individual monograph.
5.2 As a general guideline, it is expected that all new monographs for conventional-release capsules and tablets will contain a dissolution requirement except (i) where the solubility of the active ingredient at 37 ± 0.5° is high throughout the physiological pH range (dose/solubility volume <250 mL at pH 1.2 to 6.8); (ii) where the dissolution of the dosage form is greater than 80% in 15 minutes at pH 1.2, 4.0 and 6.8; (iii) where a relationship has been determined between disintegration and dissolution, or when disintegration has been observed to be more discriminatory than dissolution. In circumstances where conditions (i) to (iii) have been satisfied, the dissolution test may be replaced by the disintegration test for tablets and capsules, Appendix XII A1.
5.3 If challenged, a product would be expected to comply with the test for dissolution specified in the individual monograph, or if none is specified, with the Test conditions and acceptance criteria outlined in section 3.
5.4 Dissolution tests have been added to a considerable number of monographs in the British Pharmacopoeia. While the objective is to include a ‘standard’ pharmacopoeial test wherever appropriate, the circumstances for each preparation are considered individually in consultation with the manufacturers. It should be appreciated, however, that the retrospective addition of dissolution tests is not without its difficulties. The problems are most acute for those well-established preparations that are manufactured by a wide range of companies, each with its own dissolution specification. A pragmatic approach is being taken to developing compromise test procedures in these circumstances.
6. Bioavailability and Bioequivalence
6.1 Compliance with the standard British Pharmacopoeia requirement for dissolution provides an assurance that most of the active ingredient will be dissolved in a reasonable amount of time when the preparation is subjected to mild agitation.
6.2 It should be noted that compliance with the pharmacopoeial dissolution test does not by itself guarantee bioavailability and is not necessarily an adequate basis for judging bioequivalence between preparations. Preparations having similar dissolution characteristics may not be bioequivalent and vice versa.
7. Application of harmonised dissolution limits (“Q” values)
7.1 Since the harmonisation of the dissolution test through the Pharmacopoeial Discussion Group and the adoption of harmonised dissolution criteria (“Q values”) between the European, United States and Japanese Pharmacopoeias, the BP Commission has received many queries regarding the application of limits to dissolution tests.
7.2 As an aid to analysts several worked examples are provided as an illustrative guide below.
7.3 An immediate-release tablet formulation has a limit of Q = 75%. Six units are tested.
Unit | Result (% of stated amount) |
1 | 83 |
2 | 87 |
3 | 80 |
4 | 88 |
5 | 90 |
6 | 82 |
In order to pass S1, all units must be greater than or equal to (Q + 5)% (i.e. 80%). Hence, the above batch passes at S1.
7.4 An immediate-release tablet formulation has a limit of Q = 75%. Six units are tested.
Unit | Result (% of stated amount) |
1 | 77 |
2 | 87 |
3 | 70 |
4 | 88 |
5 | 90 |
6 | 82 |
Since units 1 and 3 are less than (Q+5)% (i.e. 80%), the batch fails at S1. A further six units must then be tested.
Unit | Result (% of stated amount) |
7 | 78 |
8 | 85 |
9 | 79 |
10 | 84 |
11 | 87 |
12 | 85 |
In order to pass S2, the mean of twelve units must be greater than or equal to Q and no unit can be less than (Q – 15)% (i.e. 60%). The mean of the twelve units is 83% so this passes at S2 and, units 1 and 3, whilst failing at S1, are not less than (Q – 15)%, so this batch passes at S2.
7.4 An immediate-release tablet formulation has a limit of Q = 75%. Six units are tested.
Unit | Result (% of stated amount) |
1 | 77 |
2 | 87 |
3 | 70 |
4 | 88 |
5 | 76 |
6 | 79 |
Since units 1, 3, 5 and 6 are less than (Q+5)% (i.e. 80%), the batch fails at S1. A further six units must then be tested
Unit | Result (% of stated amount) |
7 | 73 |
8 | 85 |
9 | 79 |
10 | 84 |
11 | 87 |
12 | 58 |
Because unit 12 is less than (Q – 15)% (i.e. 60%) the batch fails at S2. A further twelve units must then be tested.
Unit | Result (% of stated amount) |
13 | 79 |
14 | 83 |
15 | 67 |
16 | 84 |
17 | 80 |
18 | 82 |
19 | 78 |
20 | 85 |
21 | 84 |
22 | 81 |
23 | 83 |
24 | 88 |
In order to pass S3, the mean of 24 units must be greater than or equal to Q , not more than two units can be less than (Q – 15)% (i.e. 60%) and no unit can be less than (Q – 25)% (i.e. 50%). The mean of the 24 units is 80% so this passes at S3. Only unit 12 is less than (Q – 15)% and none are less than (Q – 25)% so this batch passes at S3.
7.6 An immediate-release capsule formulation has a limit of Q = 75%. Six units are tested.
Unit | Result (% of stated amount) |
1 | 83 |
2 | 82 |
3 | 80 |
4 | 79 |
5 | 49 |
6 | 77 |
Since unit 5 is less than (Q – 25)% (i.e. 50%), the batch would fail at S3. Testing further units will not enable this batch to pass the test.
7.7 An immediate-release capsule formulation has a limit of Q = 75%. 24 units have been tested.
Unit | Result (% of stated amount) |
1 | 76 |
2 | 82 |
3 | 80 |
4 | 79 |
5 | 70 |
6 | 77 |
7 | 65 |
8 | 69 |
9 | 77 |
10 | 73 |
11 | 68 |
12 | 71 |
13 | 79 |
14 | 65 |
15 | 70 |
16 | 72 |
17 | 67 |
18 | 83 |
19 | 63 |
20 | 68 |
21 | 74 |
22 | 71 |
23 | 75 |
24 | 70 |
All the units are greater than (Q – 15)% (i.e. 60%), however; because the mean (73%) is less than Q, this batch fails at S3.